

Whole-exome sequencing identification of novel DNAH5 mutations in a young patient with primary ciliary dyskinesia
- PMID: 27779714
- PMCID: PMC5355724
- DOI: 10.3892/mmr.2016.5871
Whole-exome sequencing identification of novel DNAH5 mutations in a young patient with primary ciliary dyskinesia
Abstract
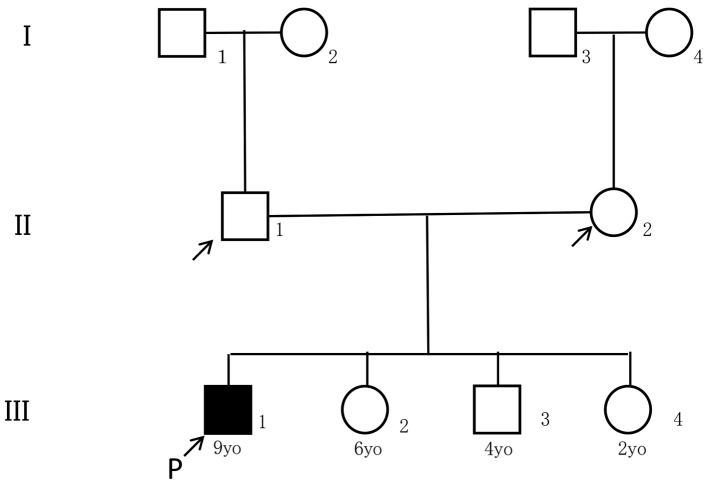
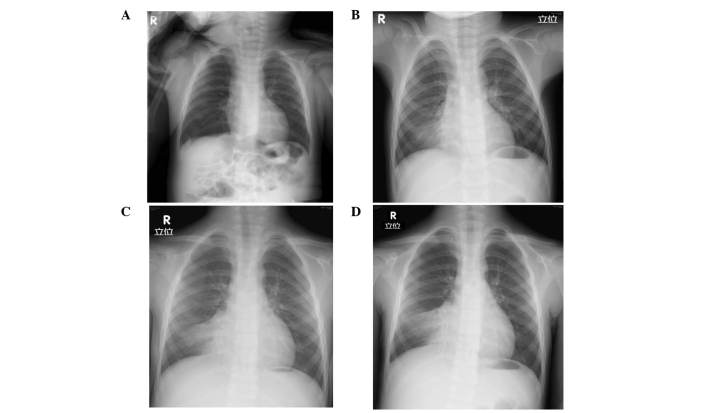
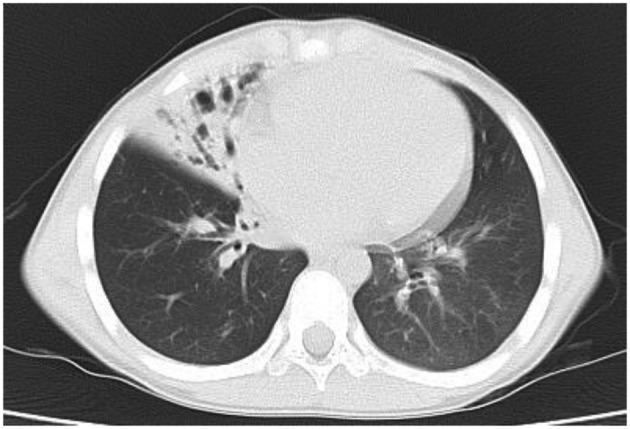
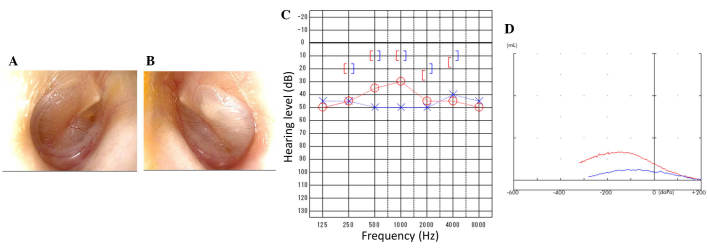
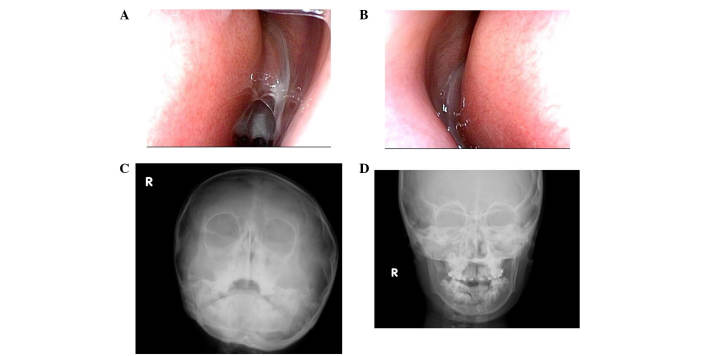
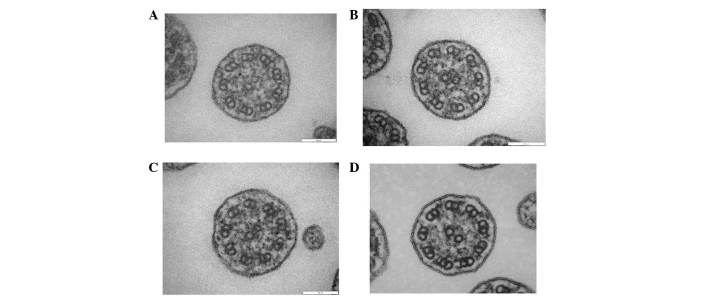
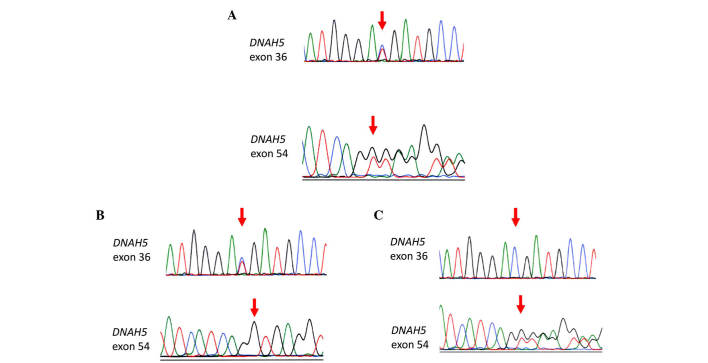
Primary ciliary dyskinesia (PCD) is a rare genetic disorder caused by structural and/or functional impairment of cilia throughout the whole body. Early diagnosis of PCD is important for the prevention of long‑term sequelae, however early diagnosis is a challenge due to the phenotypic heterogeneity of PCD. In the current study, the patient with PCD was diagnosed at nine years old following several efforts to control intractable airway symptoms. The patient experienced a chronic productive cough beginning in early childhood and had multiple episodes of pneumonia and otitis media with effusion and sinusitis. No situs inversus or other heterotaxias were reported. Serial chest X‑rays exhibited persistent atelectasis and bronchiectasis in the right middle lobe. When the patient was nine years old, electron microscopy of his cilia and genetic analysis were conducted. Electron microscopy of a biopsy specimen from the nasal mucosa indicated loss of the outer dynein arms. Whole‑exome analysis of the genome demonstrated the presence of compound heterozygous mutations in DNAH5: NM_001369.2:c.5983C>T, p.Arg1995X in exon 36 and NM_001369.2:c.9101delG, p.Gly3034ValfsX22 in exon 54; neither of which have been previously reported in the literature in a Japanese patient. Notably, this case is, to the best of our knowledge, the first reported case of PCD caused by the DNAH5 mutation in a Japanese patient.
Figures
Similar articles
-
A Japanese Case of Primary Ciliary Dyskinesia with DNAH5 Mutations.Intern Med. 2019 Aug 15;58(16):2383-2386. doi: 10.2169/internalmedicine.1961-18. Epub 2019 May 22. Intern Med. 2019. PMID: 31118369 Free PMC article.
-
Novel compound heterozygous mutations of DNAH5 identified in a pediatric patient with Kartagener syndrome: case report and literature review.BMC Pulm Med. 2021 Aug 14;21(1):263. doi: 10.1186/s12890-021-01586-4. BMC Pulm Med. 2021. PMID: 34391405 Free PMC article. Review.
-
[Clinical characteristics of primary ciliary dyskinesia].Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2014 Feb;49(2):115-20. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2014. PMID: 24742509 Chinese.
-
Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia.Am J Respir Crit Care Med. 2005 Jun 15;171(12):1343-9. doi: 10.1164/rccm.200411-1583OC. Epub 2005 Mar 4. Am J Respir Crit Care Med. 2005. PMID: 15750039 Free PMC article.
-
[Cilia ultrastructural and gene variation of primary ciliary dyskinesia: report of three cases and literatures review].Zhonghua Er Ke Za Zhi. 2018 Feb 2;56(2):134-137. doi: 10.3760/cma.j.issn.0578-1310.2018.02.012. Zhonghua Er Ke Za Zhi. 2018. PMID: 29429202 Review. Chinese.
Cited by
-
Diagnosis of Primary Ciliary Dyskinesia via Whole Exome Sequencing and Histologic Findings.Yonsei Med J. 2024 Jan;65(1):48-54. doi: 10.3349/ymj.2023.0238. Yonsei Med J. 2024. PMID: 38154480 Free PMC article.
-
A Japanese Case of Primary Ciliary Dyskinesia with DNAH5 Mutations.Intern Med. 2019 Aug 15;58(16):2383-2386. doi: 10.2169/internalmedicine.1961-18. Epub 2019 May 22. Intern Med. 2019. PMID: 31118369 Free PMC article.
-
Dual-allele heterozygous mutation of DNAH5 gene in a boy with primary ciliary dyskinesia: A case report.Medicine (Baltimore). 2023 Dec 29;102(52):e36271. doi: 10.1097/MD.0000000000036271. Medicine (Baltimore). 2023. PMID: 38206729 Free PMC article.
-
Characteristic genetic spectrum of primary ciliary dyskinesia in Japanese patients and global ethnic heterogeneity: population-based genomic variation database analysis.J Hum Genet. 2023 Jul;68(7):455-461. doi: 10.1038/s10038-023-01142-4. Epub 2023 Mar 2. J Hum Genet. 2023. PMID: 36864285
-
Primary ciliary dyskinesia: mechanisms and management.Appl Clin Genet. 2017 Sep 19;10:67-74. doi: 10.2147/TACG.S127129. eCollection 2017. Appl Clin Genet. 2017. PMID: 29033599 Free PMC article. Review.
References
-
- Afzelius BA, Mossberg B. Immotile-cilia syndrome (primary ciliary dyskinesia), including Kartagener syndrome. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 1995. pp. 3943–3954.
-
- Sommer JU, Schäfer K, Omran H, Olbrich H, Wallmeier J, Blum A, Hörmann K, Stuck BA. ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co-morbidities. Eur Arch Otorhinolaryngol. 2011;268:383–388. doi: 10.1007/s00405-010-1341-9. - DOI - PubMed
-
- Rubin BK. Immotile cilia syndrome (primary ciliary dyskinesia) and inflammatory lung disease. Clin Chest Med. 1988;9:657–668. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
