

Protein 4.1R Influences Myogenin Protein Stability and Skeletal Muscle Differentiation
- PMID: 27780863
- PMCID: PMC5207257
- DOI: 10.1074/jbc.M116.761296
Protein 4.1R Influences Myogenin Protein Stability and Skeletal Muscle Differentiation
Abstract
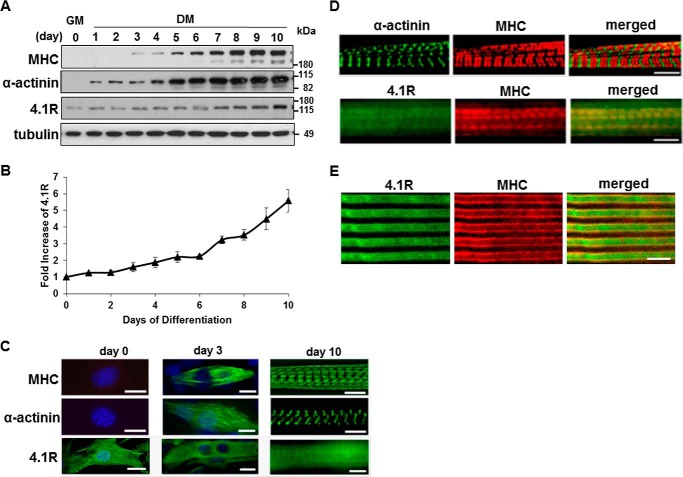
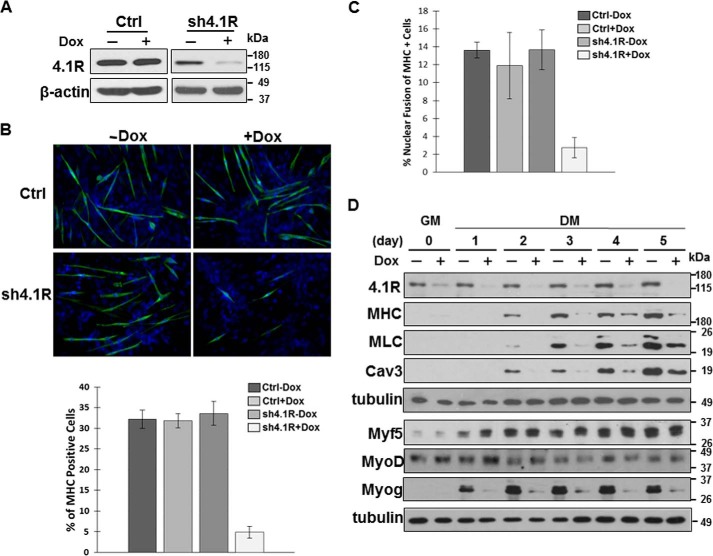
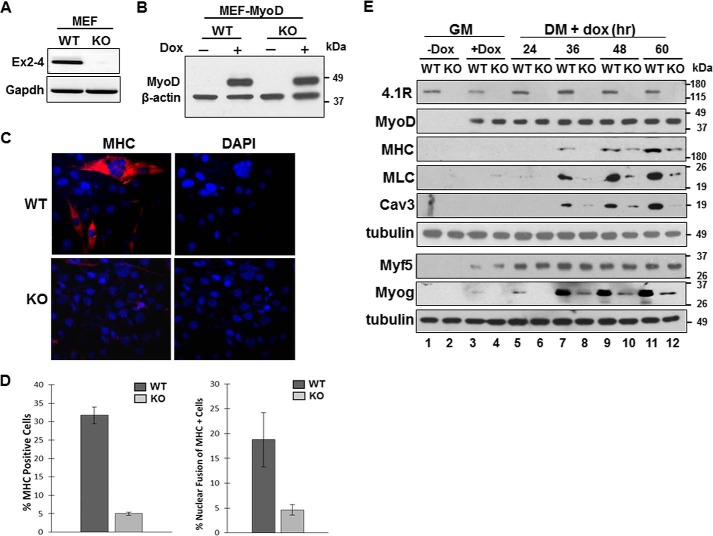
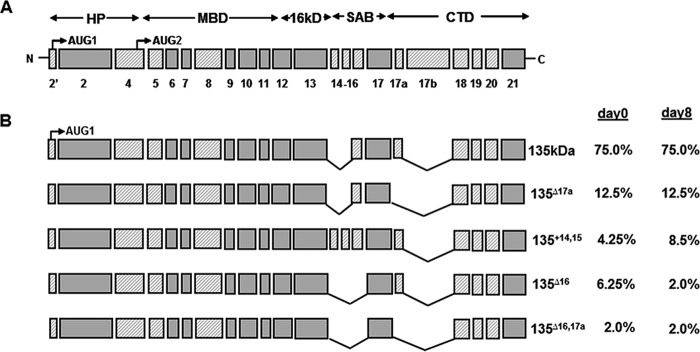
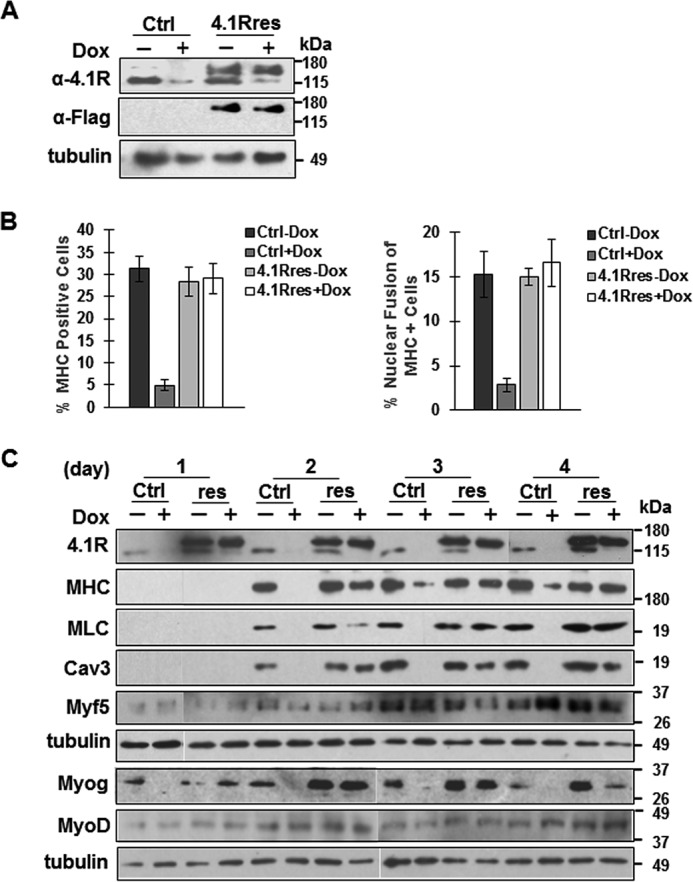
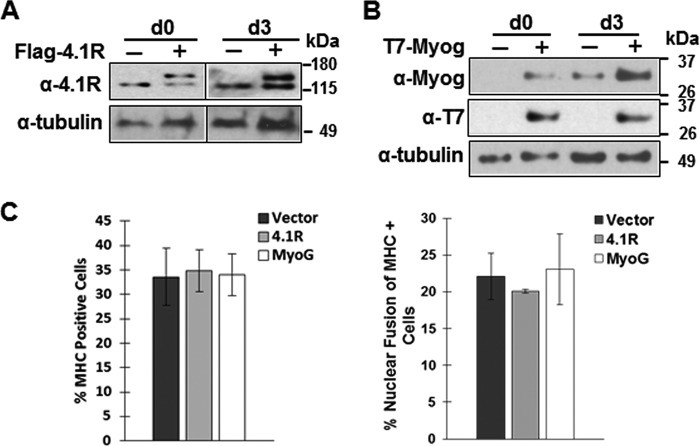
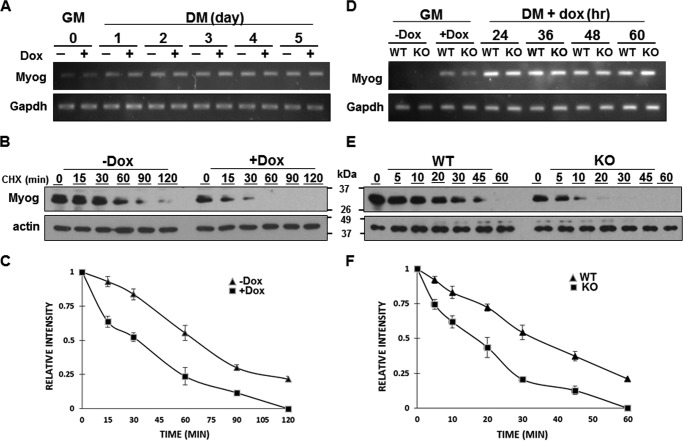
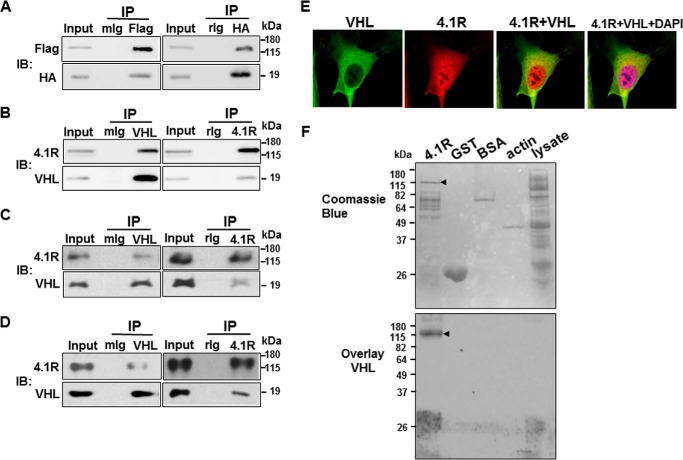
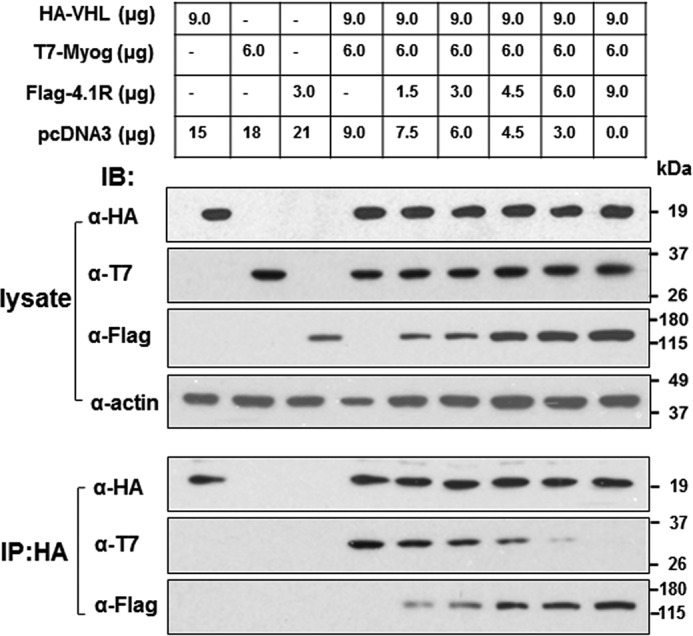
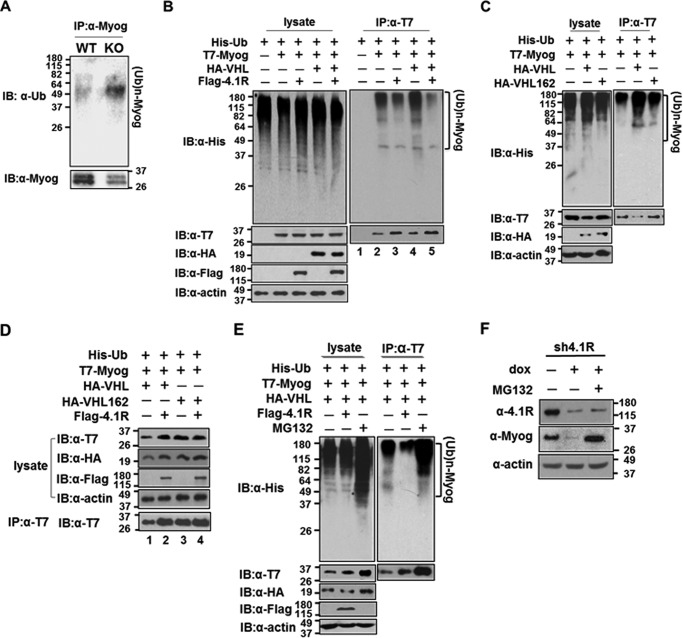
Protein 4.1R (4.1R) isoforms are expressed in both cardiac and skeletal muscle. 4.1R is a component of the contractile apparatus. It is also associated with dystrophin at the sarcolemma in skeletal myofibers. However, the expression and function of 4.1R during myogenesis have not been characterized. We now report that 4.1R expression increases during C2C12 myoblast differentiation into myotubes. Depletion of 4.1R impairs skeletal muscle differentiation and is accompanied by a decrease in the levels of myosin heavy and light chains and caveolin-3. Furthermore, the expression of myogenin at the protein, but not mRNA, level is drastically decreased in 4.1R knockdown myocytes. Similar results were obtained using MyoD-induced differentiation of 4.1R-/- mouse embryonic fibroblast cells. von Hippel-Lindau (VHL) protein is known to destabilize myogenin via the ubiquitin-proteasome pathway. We show that 4.1R associates with VHL and, when overexpressed, reverses myogenin ubiquitination and stability. This suggests that 4.1R may influence myogenesis by preventing VHL-mediated myogenin degradation. Together, our results define a novel biological function for 4.1R in muscle differentiation and provide a molecular mechanism by which 4.1R promotes myogenic differentiation.
Keywords: E3 ubiquitin ligase; myogenesis; myogenin; protein 4.1R; protein degradation; protein stability; ubiquitylation (ubiquitination); von Hippel Lindau.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Lallena M. J., Martínez C., Valcárcel J., and Correas I. (1998) Functional association of nuclear protein 4.1 with pre-mRNA splicing factors. J. Cell Sci. 111, 1963–1971 - PubMed
-
- Mattagajasingh S. N., Huang S. C., Hartenstein J. S., and Benz E. J. Jr. (2000) Characterization of the interaction between protein 4.1R and ZO-2: a possible link between the tight junction and the actin cytoskeleton. J. Biol. Chem. 275, 30573–30585 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
