

Understanding Genotypes and Phenotypes in Epileptic Encephalopathies
- PMID: 27781027
- PMCID: PMC5073622
- DOI: 10.1159/000448530
Understanding Genotypes and Phenotypes in Epileptic Encephalopathies
Abstract
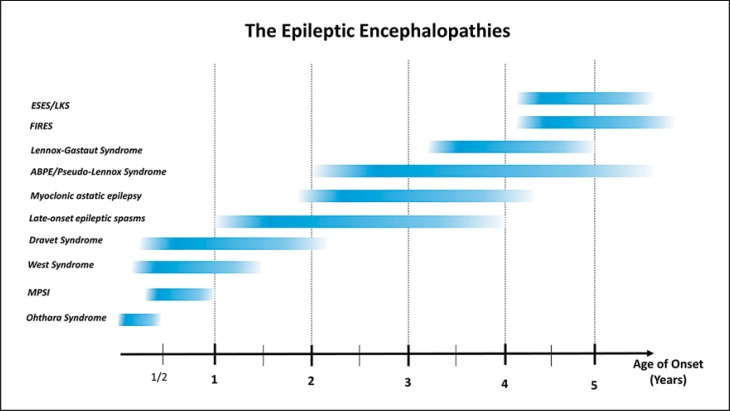
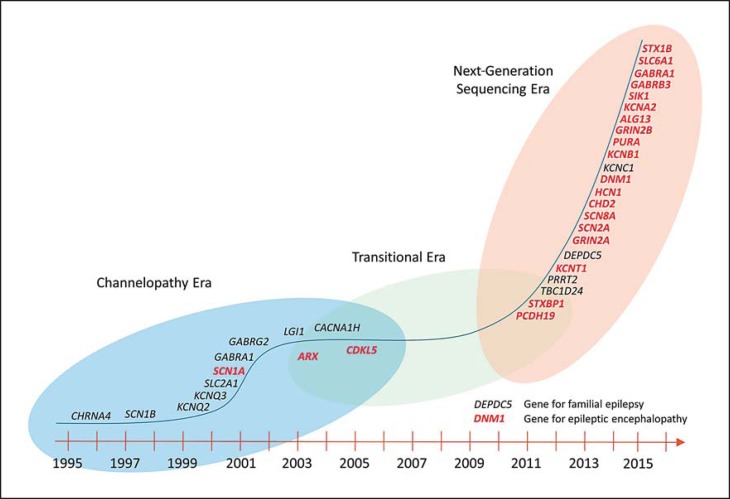
Epileptic encephalopathies are severe often intractable seizure disorders where epileptiform abnormalities contribute to a progressive disturbance in brain function. Often, epileptic encephalopathies start in childhood and are accompanied by developmental delay and various neurological and non-neurological comorbidities. In recent years, this concept has become virtually synonymous with a group of severe childhood epilepsies including West syndrome, Lennox-Gastaut syndrome, Dravet syndrome, and several other severe childhood epilepsies for which genetic factors are increasingly recognized. In the last 5 years, the field has seen a virtual explosion of gene discovery, raising the number of bona fide genes and possible candidate genes for epileptic encephalopathies to more than 70 genes, explaining 20-25% of all cases with severe early-onset epilepsies that had otherwise no identifiable causes. This review will focus on the phenotypic variability as a characteristic aspect of genetic epilepsies. For many genetic epilepsies, the phenotypic presentation can be broad, even in patients with identical genetic alterations. Furthermore, patients with different genetic etiologies can have seemingly similar clinical presentations, such as in Dravet syndrome. While most patients carry mutations in SCN1A, similar phenotypes can be seen in patients with mutations in PCDH19, CHD2, SCN8A, or in rare cases GABRA1 and STXBP1. In addition to the genotypic and phenotypic heterogeneity, both benign phenotypes and severe encephalopathies have been recognized in an increasing number of genetic epilepsies, raising the question whether these conditions represent a fluid continuum or distinct entities.
Keywords: Epileptic encephalopathy; Genotypic heterogeneity; Next-generation sequencing; Phenotypic heterogeneity; Whole-exome sequencing.
Figures
References
-
- Bayat A, Hjalgrim H, Møller RS. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22,000: a population-based study from 2004 to 2009. Epilepsia. 2015;56:e36–39. - PubMed
-
- Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–557. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
