

Injury site-specific targeting of complement inhibitors for treating stroke
- PMID: 27782326
- PMCID: PMC5098802
- DOI: 10.1111/imr.12470
Injury site-specific targeting of complement inhibitors for treating stroke
Abstract
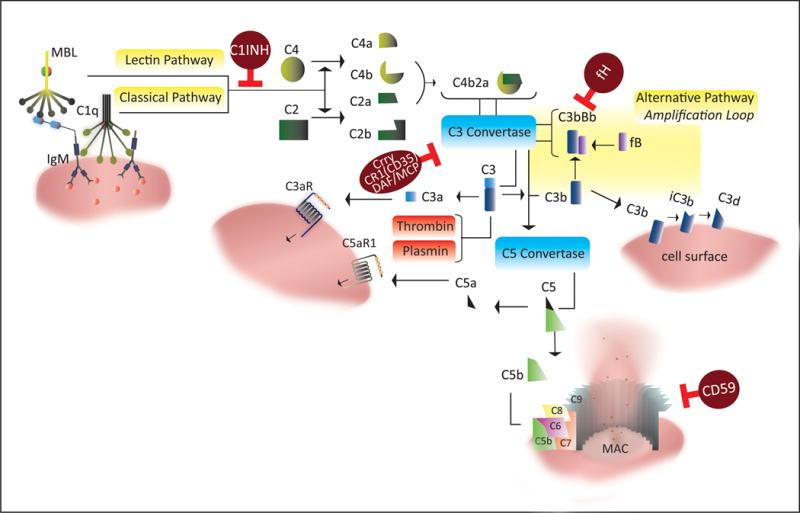
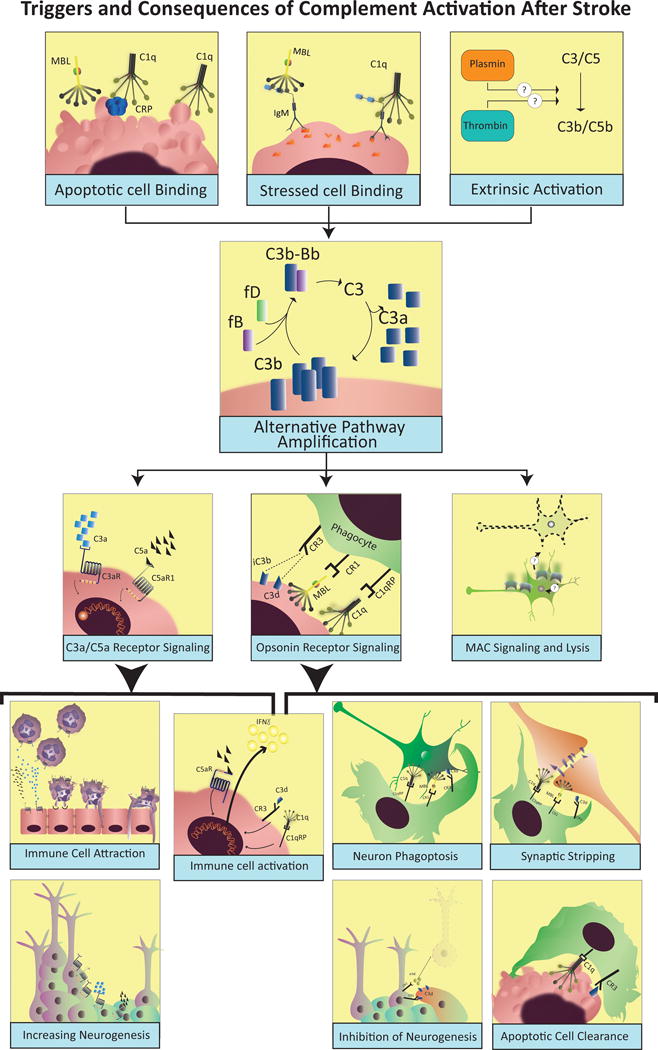
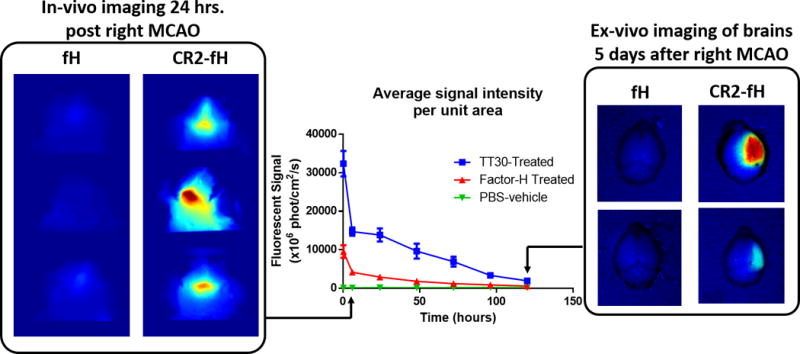
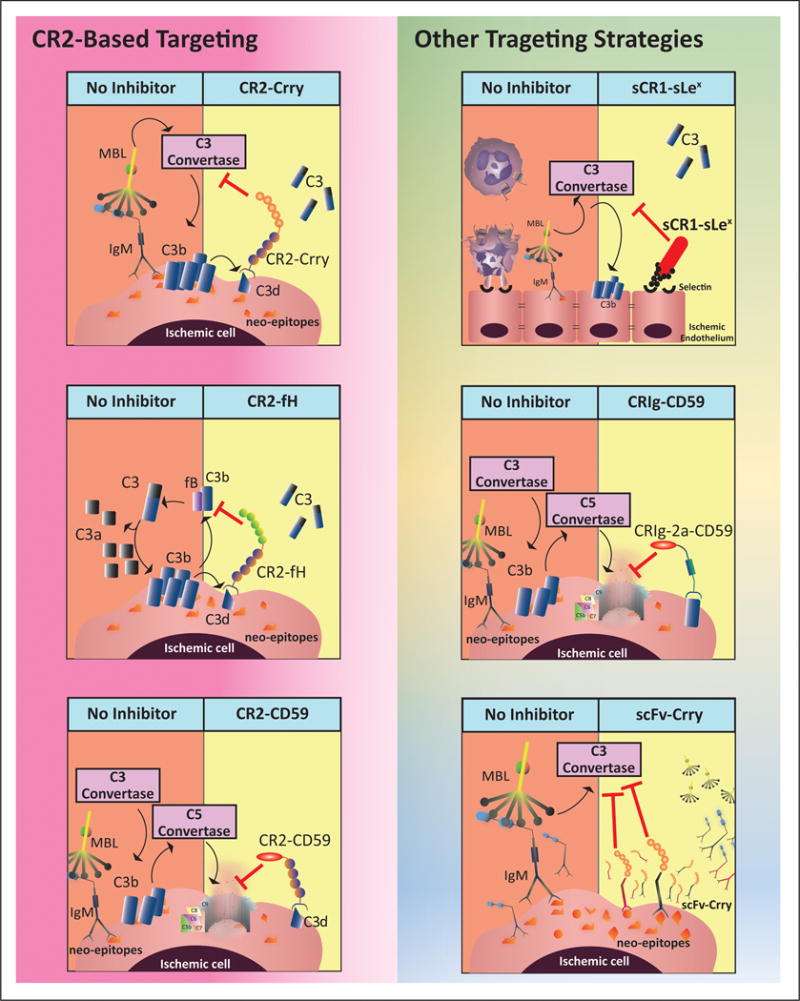
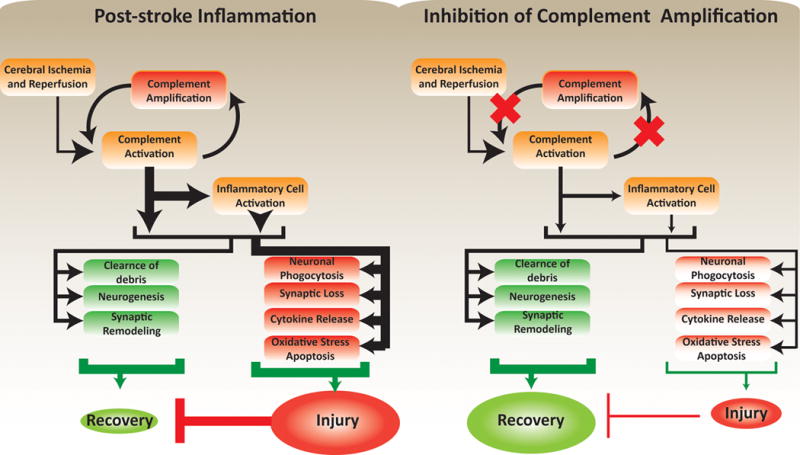
Cumulative evidence indicates a role for the complement system in both pathology and recovery after ischemic stroke. Here, we review the current understanding of the dual role of complement in poststroke injury and recovery, and discuss the challenges of anti-complement therapies. Most complement directed therapeutics currently under investigation or development systemically inhibit the complement system, but since complement is important for immune surveillance and is involved in various homeostatic activities, there are potential risks associated with systemic inhibition. Depending on the target within the complement pathway, other concerns are high concentrations of inhibitor required, low efficacy and poor bioavailability. To overcome these limitations, approaches to target complement inhibitors to specific sites have been investigated. Here, we discuss targeting strategies, with a focus on strategies developed in our lab, to specifically localize complement inhibition to sites of tissue injury and complement activation, and in particular to the postischemic brain. We discuss various injury site-specific targeted complement inhibitors as potential therapeutic agents for the treatment of ischemic stroke treatment, as well as their use as investigative tools for probing complement-dependent pathophysiological processes.
Keywords: complement system; inflammation; stroke; stroke therapeutics; tissue targeting.
© 2016 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors have no conflicts of interest
Figures
References
-
- Mozaffarian Dariush, et al. Executive Summary: Heart Disease and Stroke Statistics—2016 Update A Report From the American Heart Association. Circulation. 2016;133.4:447–454. APA. - PubMed
-
- Kochanek K, Murphy S, Xu J, Arias E. Mortality in the United States, 2013. Hyattsville, MD: National Center for Health Statistics; 2014. (NCHS data brief, no. 178). 2014. - PubMed
-
- Murray CJ, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. - PubMed
-
- Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42:2351–2355. - PubMed
-
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–1587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
