

1970s and 'Patient 0' HIV-1 genomes illuminate early HIV/AIDS history in North America
- PMID: 27783600
- PMCID: PMC5257289
- DOI: 10.1038/nature19827
1970s and 'Patient 0' HIV-1 genomes illuminate early HIV/AIDS history in North America
Abstract
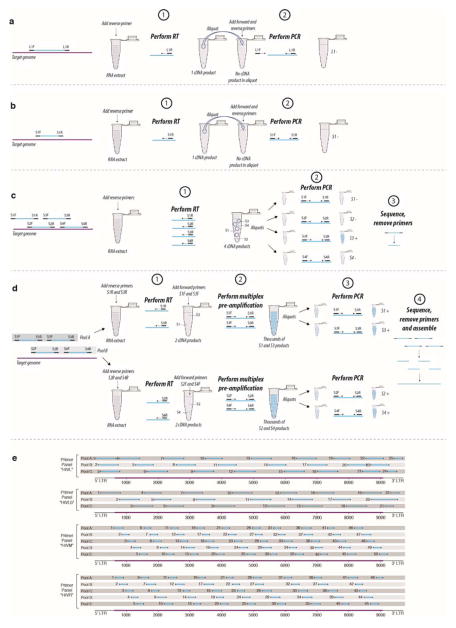
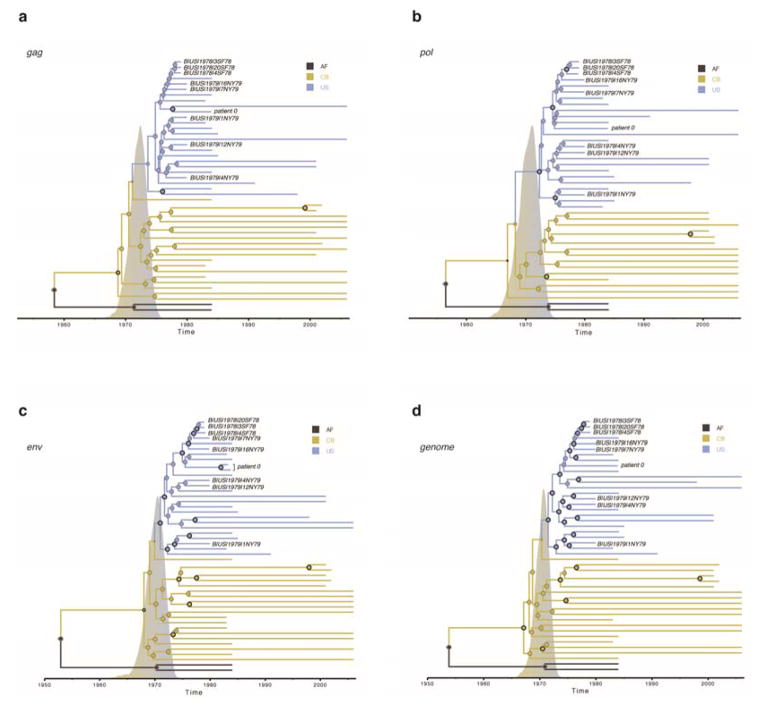
The emergence of HIV-1 group M subtype B in North American men who have sex with men was a key turning point in the HIV/AIDS pandemic. Phylogenetic studies have suggested cryptic subtype B circulation in the United States (US) throughout the 1970s and an even older presence in the Caribbean. However, these temporal and geographical inferences, based upon partial HIV-1 genomes that postdate the recognition of AIDS in 1981, remain contentious and the earliest movements of the virus within the US are unknown. We serologically screened >2,000 1970s serum samples and developed a highly sensitive approach for recovering viral RNA from degraded archival samples. Here, we report eight coding-complete genomes from US serum samples from 1978-1979-eight of the nine oldest HIV-1 group M genomes to date. This early, full-genome 'snapshot' reveals that the US HIV-1 epidemic exhibited extensive genetic diversity in the 1970s but also provides strong evidence for its emergence from a pre-existing Caribbean epidemic. Bayesian phylogenetic analyses estimate the jump to the US at around 1970 and place the ancestral US virus in New York City with 0.99 posterior probability support, strongly suggesting this was the crucial hub of early US HIV/AIDS diversification. Logistic growth coalescent models reveal epidemic doubling times of 0.86 and 1.12 years for the US and Caribbean, respectively, suggesting rapid early expansion in each location. Comparisons with more recent data reveal many of these insights to be unattainable without archival, full-genome sequences. We also recovered the HIV-1 genome from the individual known as 'Patient 0' (ref. 5) and found neither biological nor historical evidence that he was the primary case in the US or for subtype B as a whole. We discuss the genesis and persistence of this belief in the light of these evolutionary insights.
Conflict of interest statement
The authors declare no competing financial interests. A patent, “Methods and systems for RNA or DNA detection and sequencing” (U.S. patent application 62/325,320), has been filed with the U.S. Patent Office. It will be used to facilitate the nonexclusive licensing of this methodology.
Figures










Comment in
-
How researchers cleared the name of HIV Patient Zero.Nature. 2016 Oct 27;538(7626):428. doi: 10.1038/538428a. Nature. 2016. PMID: 27786222 No abstract available.
-
'Patient 0' and the Origin of HIV/AIDS in America.Trends Microbiol. 2017 Jan;25(1):3-4. doi: 10.1016/j.tim.2016.11.005. Epub 2016 Nov 17. Trends Microbiol. 2017. PMID: 27866834
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
