

Controlling the Rate of GWAS False Discoveries
- PMID: 27784720
- PMCID: PMC5223524
- DOI: 10.1534/genetics.116.193987
Controlling the Rate of GWAS False Discoveries
Abstract
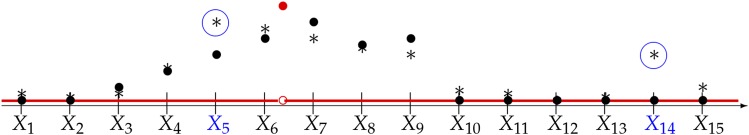
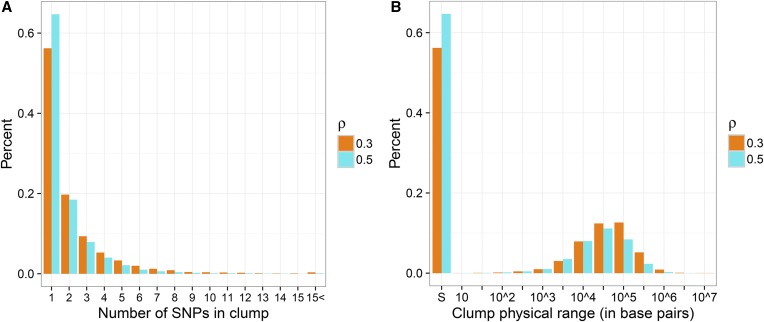
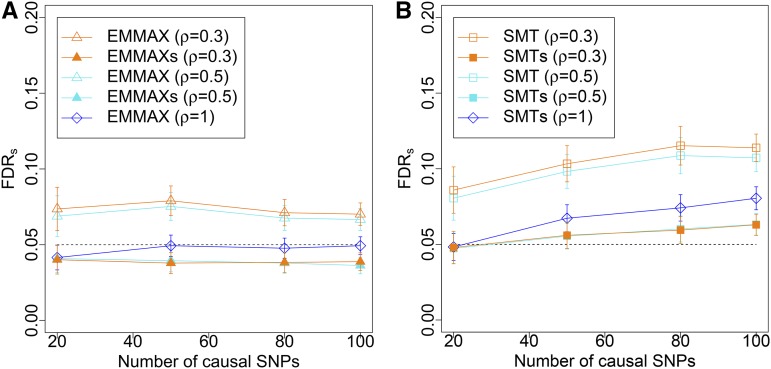
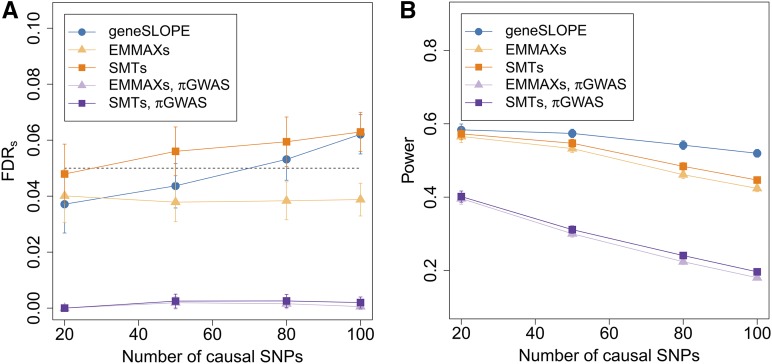
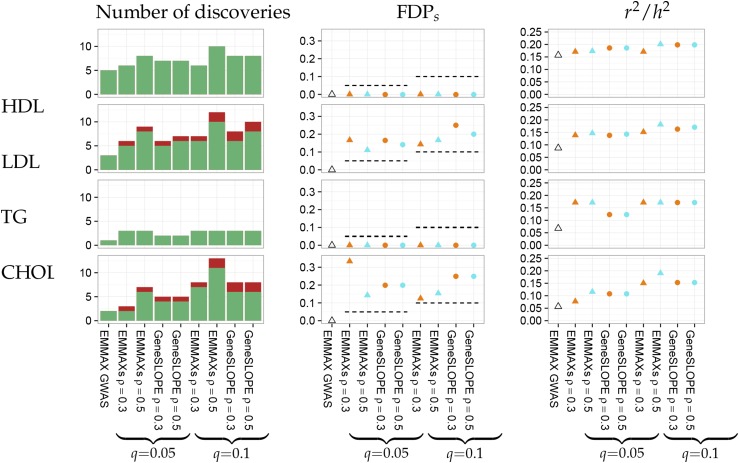
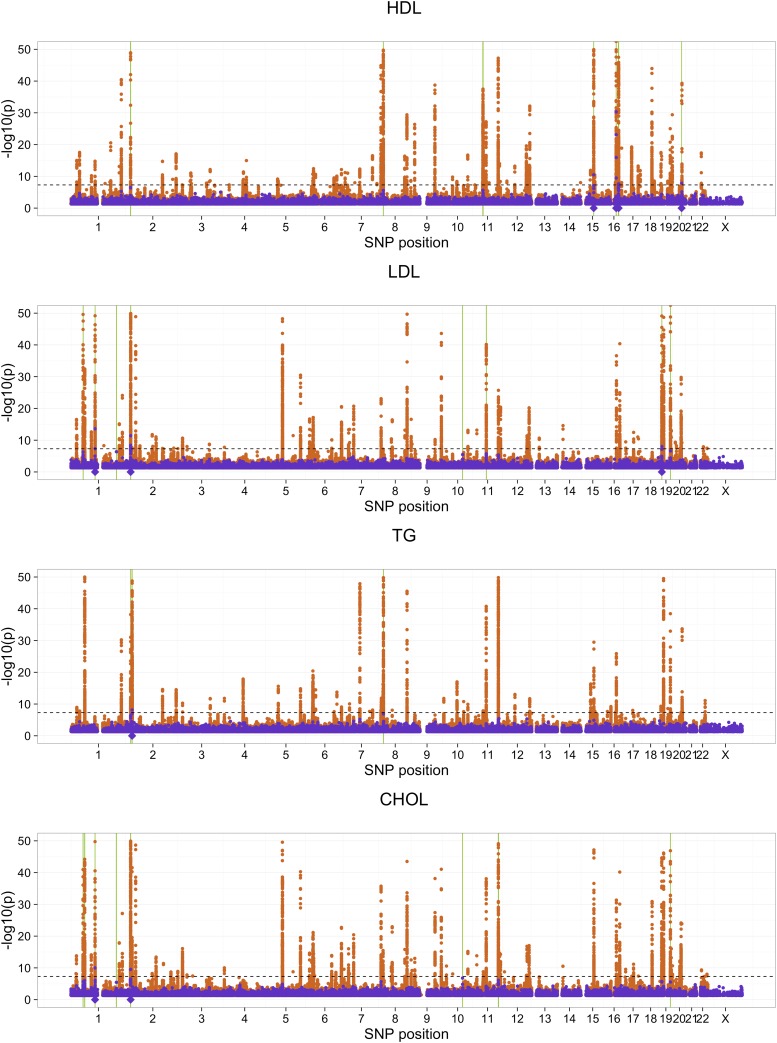
With the rise of both the number and the complexity of traits of interest, control of the false discovery rate (FDR) in genetic association studies has become an increasingly appealing and accepted target for multiple comparison adjustment. While a number of robust FDR-controlling strategies exist, the nature of this error rate is intimately tied to the precise way in which discoveries are counted, and the performance of FDR-controlling procedures is satisfactory only if there is a one-to-one correspondence between what scientists describe as unique discoveries and the number of rejected hypotheses. The presence of linkage disequilibrium between markers in genome-wide association studies (GWAS) often leads researchers to consider the signal associated to multiple neighboring SNPs as indicating the existence of a single genomic locus with possible influence on the phenotype. This a posteriori aggregation of rejected hypotheses results in inflation of the relevant FDR. We propose a novel approach to FDR control that is based on prescreening to identify the level of resolution of distinct hypotheses. We show how FDR-controlling strategies can be adapted to account for this initial selection both with theoretical results and simulations that mimic the dependence structure to be expected in GWAS. We demonstrate that our approach is versatile and useful when the data are analyzed using both tests based on single markers and multiple regression. We provide an R package that allows practitioners to apply our procedure on standard GWAS format data, and illustrate its performance on lipid traits in the North Finland Birth Cohort 66 cohort study.
Keywords: FDR; association studies; linkage disequilibrium; multiple penalized regression.
Copyright © 2017 by the Genetics Society of America.
Figures
References
-
- Abramovich F., Benjamini Y., Donoho D. L., Johnstone I. M., 2006. Adapting to unknown sparsity by controlling the false discovery rate. Ann. Stat. 34: 584–653.
-
- Alexander D. H., Lange K., 2011. Stability selection for genome-wide association. Genet. Epidemiol. 35: 722–728. - PubMed
-
- Benjamini Y., Bogomolov M., 2014. Selective inference on multiple families of hypotheses. J. R. Stat. Soc. Series B Stat. Methodol. 76: 297–318.
-
- Benjamini Y., Heller R., 2007. False discovery rates for spatial signals. J. Am. Stat. Assoc. 102: 1272–1281.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
