

Neighboring phosphoSer-Pro motifs in the undefined domain of IRAK1 impart bivalent advantage for Pin1 binding
- PMID: 27790836
- PMCID: PMC5298935
- DOI: 10.1111/febs.13943
Neighboring phosphoSer-Pro motifs in the undefined domain of IRAK1 impart bivalent advantage for Pin1 binding
Abstract
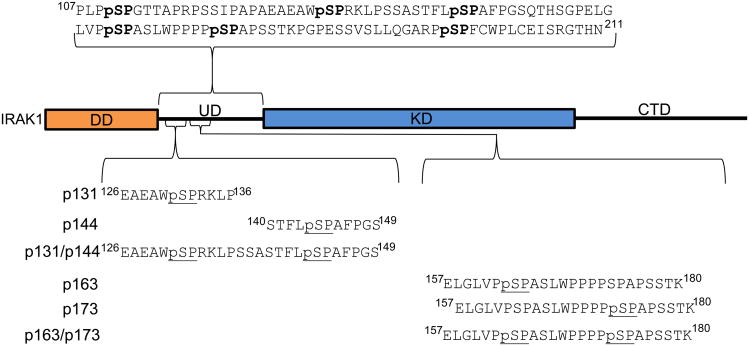
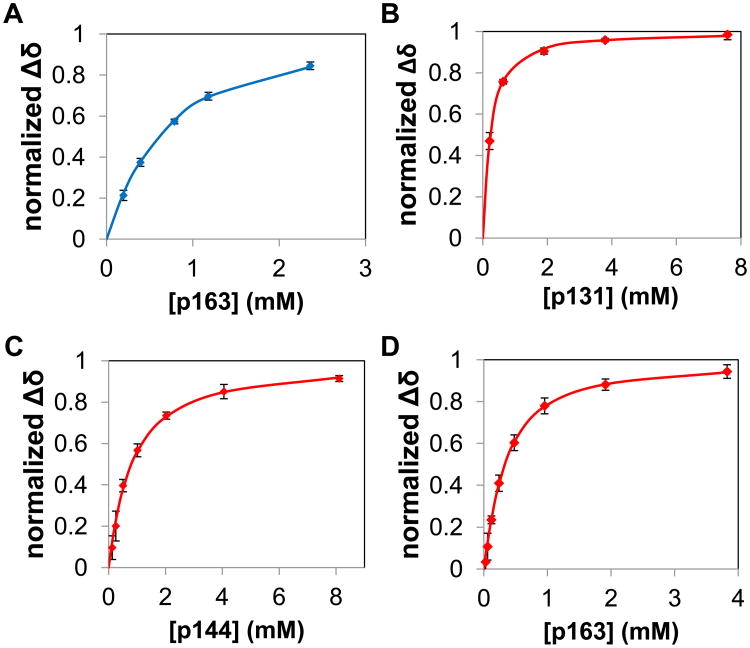
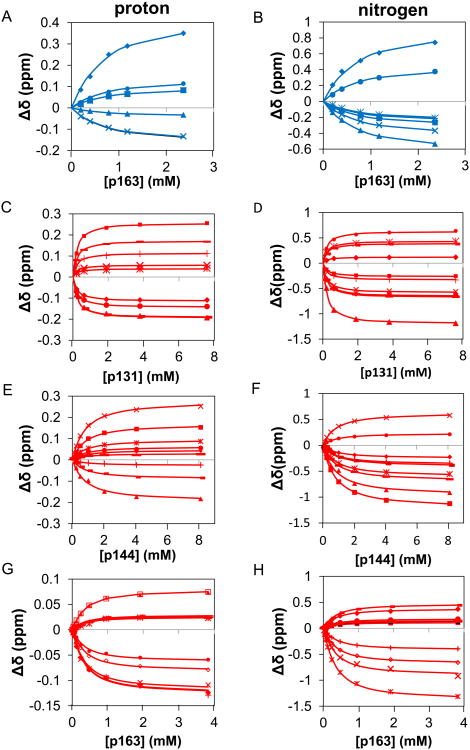
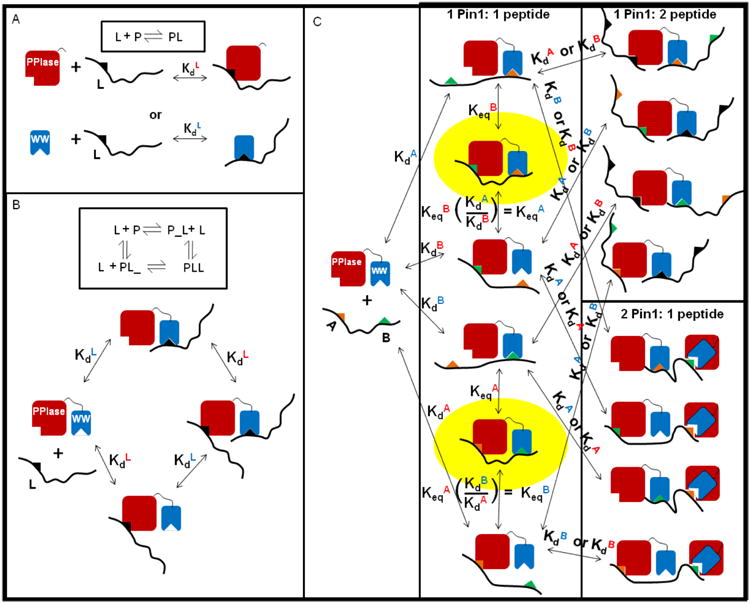
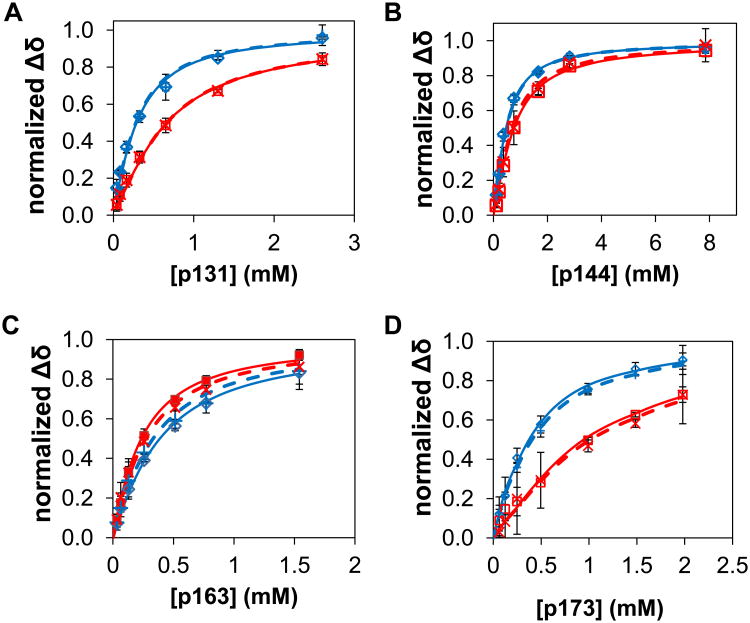
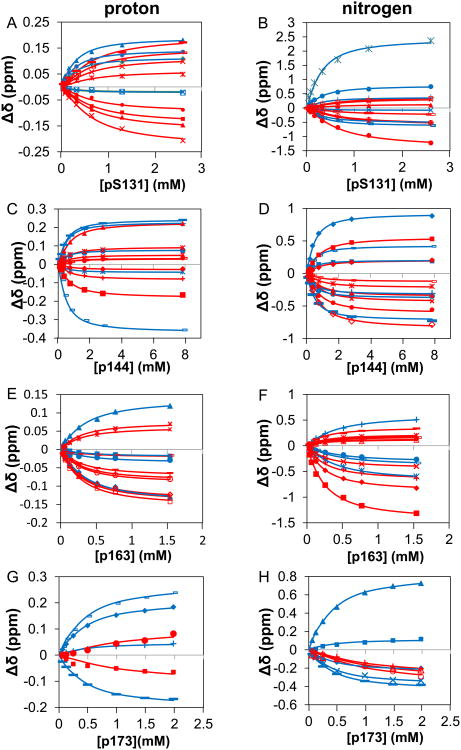
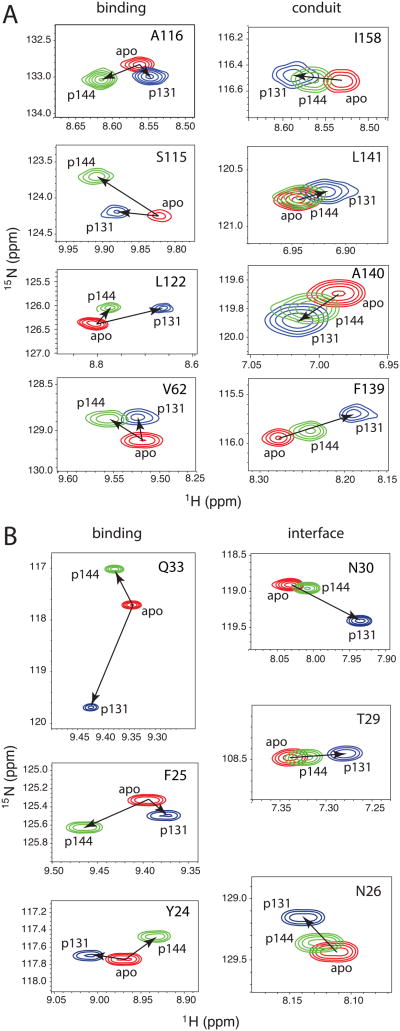
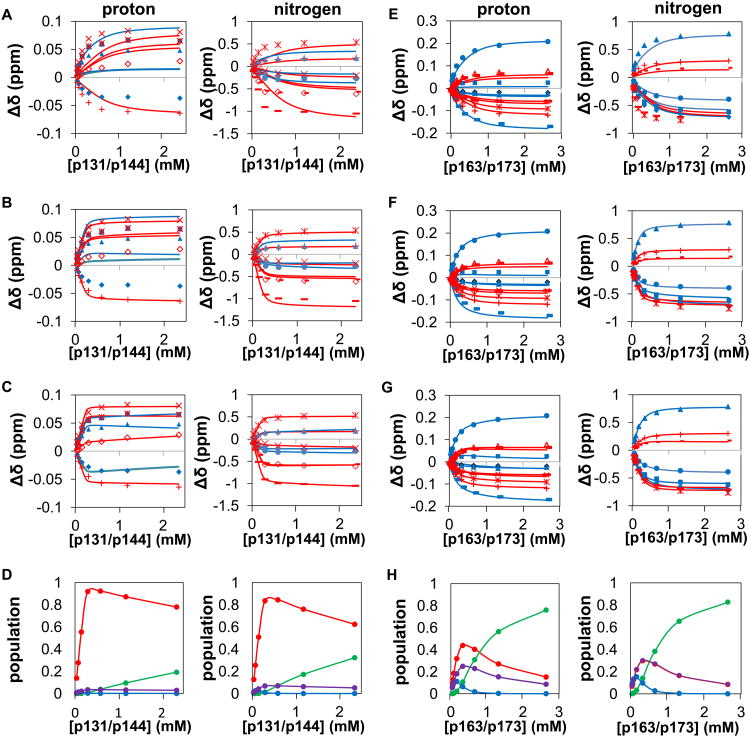
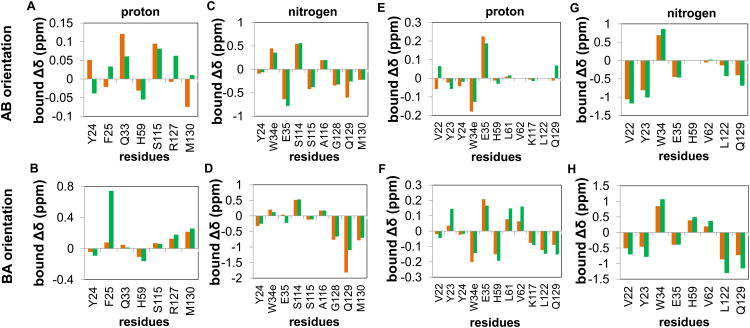
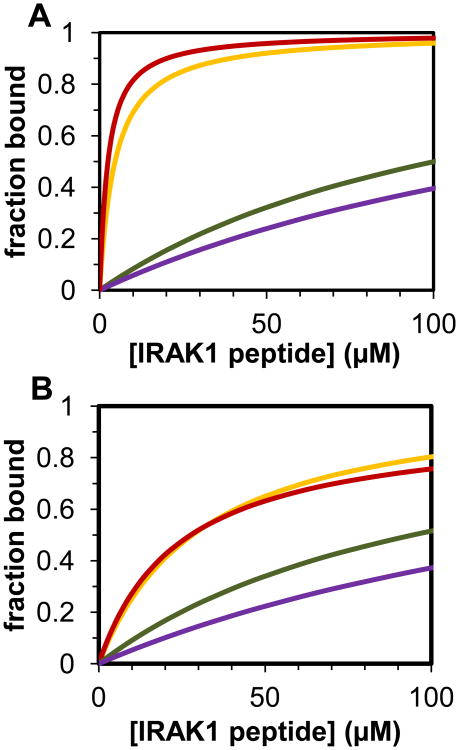
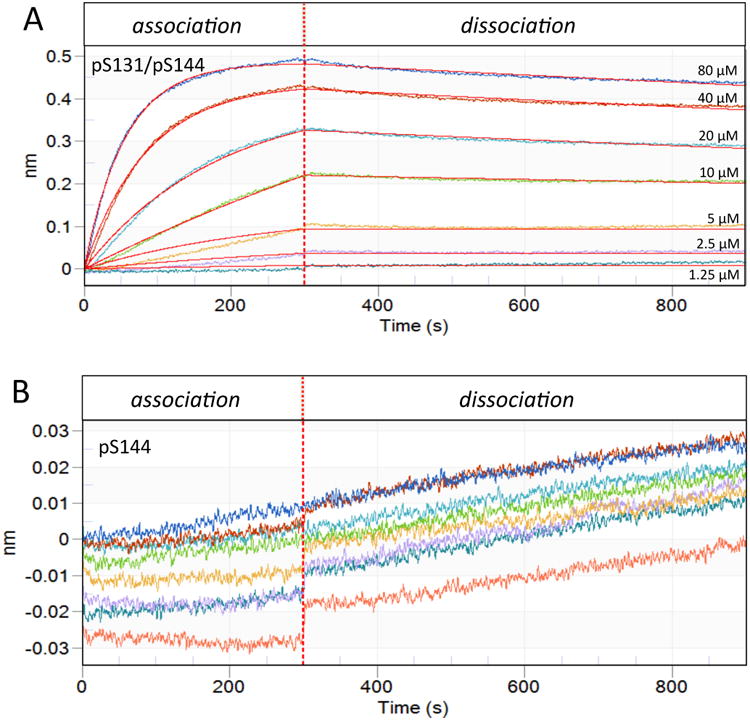
The peptidyl prolyl isomerase Pin1 has two domains that are considered to be its binding (WW) and catalytic (PPIase) domains, both of which interact with phosphorylated Ser/Thr-Pro motifs. This shared specificity might influence substrate selection, as many known Pin1 substrates have multiple sequentially close phosphoSer/Thr-Pro motifs, including the protein interleukin-1 receptor-associated kinase-1 (IRAK1). The IRAK1 undefined domain (UD) contains two sets of such neighboring motifs (Ser131/Ser144 and Ser163/Ser173), suggesting possible bivalent interactions with Pin1. Using a series of NMR titrations with 15N-labeled full-length Pin1 (Pin1-FL), PPIase, or WW domain and phosphopeptides representing the Ser131/Ser144 and Ser163/Ser173 regions of IRAK1-UD, bivalent interactions were investigated. Binding studies using singly phosphorylated peptides showed that individual motifs displayed weak affinities (> 100 μm) for Pin1-FL and each isolated domain. Analysis of dually phosphorylated peptides binding to Pin1-FL showed that inclusion of bivalent states was necessary to fit the data. The resulting complex model and fitted parameters were applied to predict the impact of bivalent states at low micromolar concentrations, demonstrating significant affinity enhancement for both dually phosphorylated peptides (3.5 and 24 μm for peptides based on the Ser131/Ser144 and Ser163/Ser173 regions, respectively). The complementary technique biolayer interferometry confirmed the predicted affinity enhancement for a representative set of singly and dually phosphorylated Ser131/Ser144 peptides at low micromolar concentrations, validating model predictions. These studies provide novel insights regarding the complexity of interactions between Pin1 and activated IRAK1, and more broadly suggest that phosphorylation of neighboring Ser/Thr-Pro motifs in proteins might provide competitive advantage at cellular concentrations for engaging with Pin1.
Keywords: NMR titration; Pin1; bivalent interaction; interleukin-1 receptor-associated kinase-1; multi-state equilibrium.
© 2016 Federation of European Biochemical Societies.
Conflict of interest statement
Dr. Lu declares that he is an inventor of Pin1 technology, which was licensed by BIDMC to Pinteon Therapeutics. Dr. Lu owns equity in, and consults for, Pinteon. His interests were reviewed and are managed by BIDMC in accordance with its conflict of interest policy.
Figures
Similar articles
-
A tethering mechanism underlies Pin1-catalyzed proline cis-trans isomerization at a noncanonical site.Proc Natl Acad Sci U S A. 2025 May 27;122(21):e2414606122. doi: 10.1073/pnas.2414606122. Epub 2025 May 19. Proc Natl Acad Sci U S A. 2025. PMID: 40388619
-
Role of phosphorylation in determining the backbone dynamics of the serine/threonine-proline motif and Pin1 substrate recognition.Biochemistry. 1998 Apr 21;37(16):5566-75. doi: 10.1021/bi973060z. Biochemistry. 1998. PMID: 9548941
-
Peptide binding induces large scale changes in inter-domain mobility in human Pin1.J Biol Chem. 2003 Jul 11;278(28):26174-82. doi: 10.1074/jbc.M300796200. Epub 2003 Apr 9. J Biol Chem. 2003. PMID: 12686540
-
Activity and Affinity of Pin1 Variants.Molecules. 2019 Dec 20;25(1):36. doi: 10.3390/molecules25010036. Molecules. 2019. PMID: 31861908 Free PMC article. Review.
-
Phosphorylation-dependent prolyl isomerization: a novel signaling regulatory mechanism.Cell Mol Life Sci. 1999 Nov 30;56(9-10):788-806. doi: 10.1007/s000180050026. Cell Mol Life Sci. 1999. PMID: 11212339 Free PMC article. Review.
Cited by
-
A tethering mechanism underlies Pin1-catalyzed proline cis-trans isomerization at a noncanonical site.Proc Natl Acad Sci U S A. 2025 May 27;122(21):e2414606122. doi: 10.1073/pnas.2414606122. Epub 2025 May 19. Proc Natl Acad Sci U S A. 2025. PMID: 40388619
-
Ligand-specific conformational change drives interdomain allostery in Pin1.Nat Commun. 2022 Aug 4;13(1):4546. doi: 10.1038/s41467-022-32340-x. Nat Commun. 2022. PMID: 35927276 Free PMC article.
-
Peptidyl-Prolyl Cis/Trans Isomerase Pin1 and Alzheimer's Disease.Front Cell Dev Biol. 2020 May 15;8:355. doi: 10.3389/fcell.2020.00355. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32500074 Free PMC article. Review.
-
Regulation of eukaryotic protein kinases by Pin1, a peptidyl-prolyl isomerase.Adv Biol Regul. 2023 Jan;87:100938. doi: 10.1016/j.jbior.2022.100938. Epub 2022 Nov 30. Adv Biol Regul. 2023. PMID: 36496344 Free PMC article.
-
Pin1 facilitates isoproterenol‑induced cardiac fibrosis and collagen deposition by promoting oxidative stress and activating the MEK1/2‑ERK1/2 signal transduction pathway in rats.Int J Mol Med. 2018 Mar;41(3):1573-1583. doi: 10.3892/ijmm.2017.3354. Epub 2017 Dec 29. Int J Mol Med. 2018. PMID: 29286102 Free PMC article.
References
-
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–16. - PubMed
-
- Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–53. - PubMed
-
- Ciarapica R, Methot L, Tang Y, Lo R, Dali R, Buscarlet M, Locatelli F, del Sal G, Rota R, Stifani S. Prolyl isomerase Pin1 and protein kinase HIPK2 cooperate to promote cortical neurogenesis by suppressing Groucho/TLE:Hes1-mediated inhibition of neuronal differentiation. Cell Death Differ. 2014;21:321–32. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
