

Next-generation sequencing technologies for detection of modified nucleotides in RNAs
- PMID: 27791472
- PMCID: PMC5699547
- DOI: 10.1080/15476286.2016.1251543
Next-generation sequencing technologies for detection of modified nucleotides in RNAs
Abstract
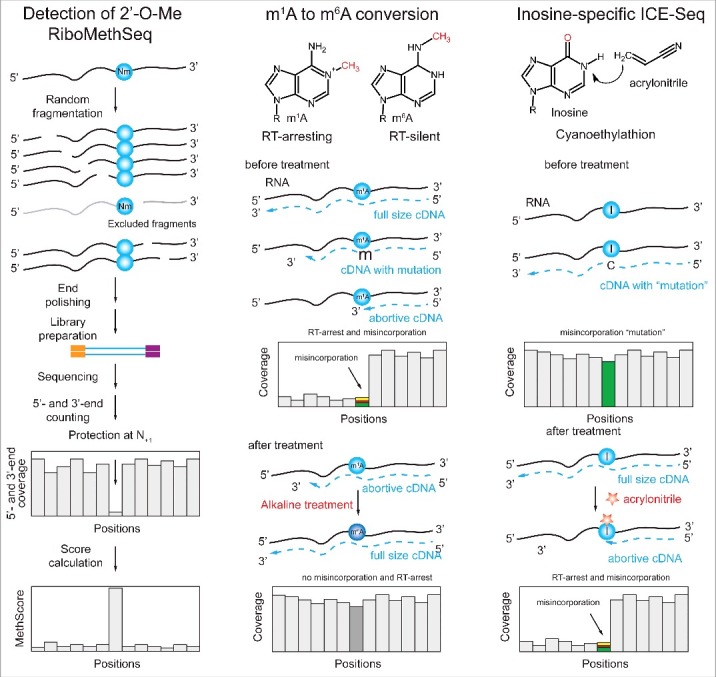
Our ability to map and quantify RNA modifications at a genome-wide scale have revolutionized our understanding of the pervasiveness and dynamic regulation of diverse RNA modifications. Recent efforts in the field have demonstrated the presence of modified residues in almost any type of cellular RNA. Next-generation sequencing (NGS) technologies are the primary choice for transcriptome-wide RNA modification mapping. Here we provide an overview of approaches for RNA modification detection based on their RT-signature, specific chemicals, antibody-dependent (Ab) enrichment, or combinations thereof. We further discuss sources of artifacts in genome-wide modification maps, and experimental and computational considerations to overcome them. The future in this field is tightly linked to the development of new specific chemical reagents, highly specific Ab against RNA modifications and use of single-molecule RNA sequencing techniques.
Keywords: Bioinformatics; RNA maturation; RNA modifications; epitranscriptome; next-generation sequencing.
Figures

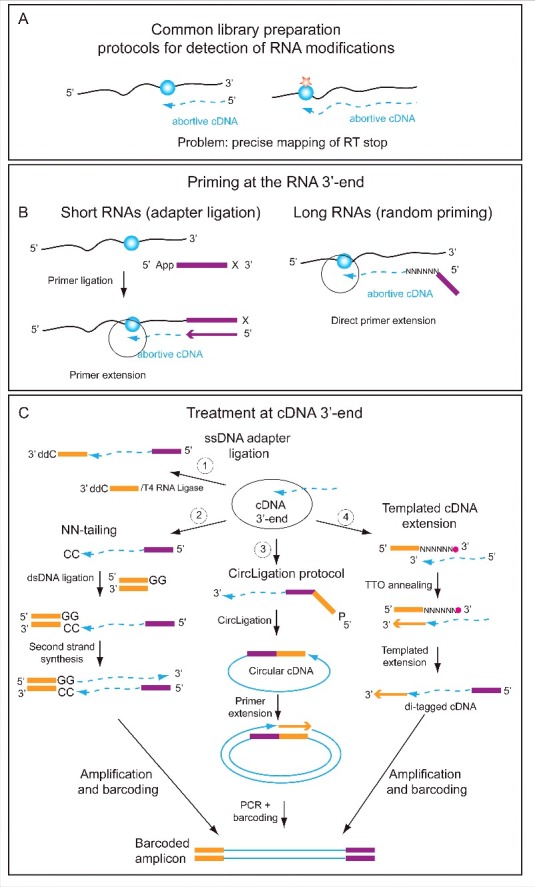
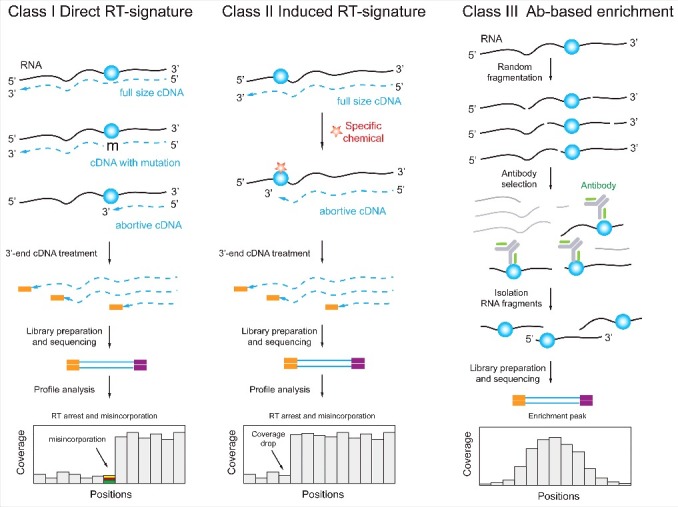
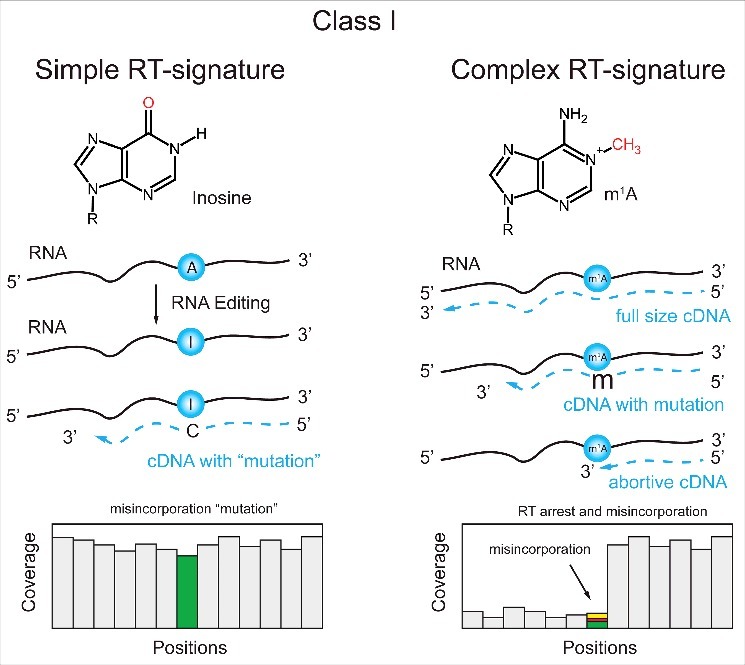
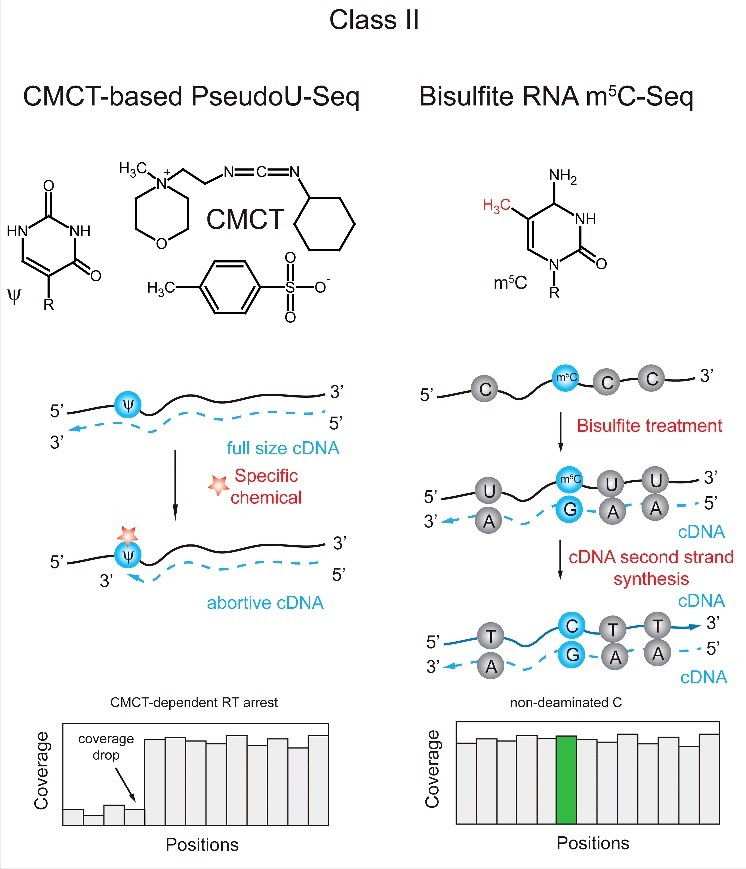


References
-
- Lee Y, Rio DC. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem 2015; 84:291-323; PMID:25784052; http://dx.doi.org/10.1146/annurev-biochem-060614-034316 - DOI - PMC - PubMed
-
- Licht K, Jantsch MF. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol 2016; 213:15-22; PMID:27044895; http://dx.doi.org/10.1083/jcb.201511041 - DOI - PMC - PubMed
-
- Wu R, Jiang D, Wang Y, Wang X. N (6)-methyladenosine (m(6)A) methylation in mRNA with A dynamic and reversible epigenetic modification. Mol Biotechnol 2016; 58:450-9; PMID:27179969; http://dx.doi.org/10.1007/s12033-016-9947-9 - DOI - PubMed
-
- Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet 2013; 29:108-15; PMID:23218460; http://dx.doi.org/10.1016/j.tig.2012.11.003 - DOI - PMC - PubMed
-
- El Yacoubi B, Bailly M, de Crécy-Lagard V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 2012; 46:69-95; PMID:22905870; http://dx.doi.org/10.1146/annurev-genet-110711-155641 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources