

Molecular dynamics simulations of aptamer-binding reveal generalized allostery in thrombin
- PMID: 27794633
- PMCID: PMC6876308
- DOI: 10.1080/07391102.2016.1254682
Molecular dynamics simulations of aptamer-binding reveal generalized allostery in thrombin
Abstract
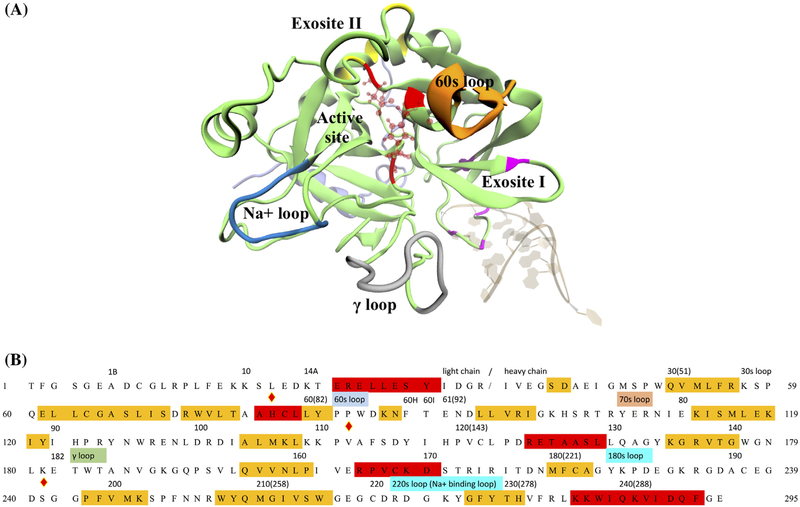
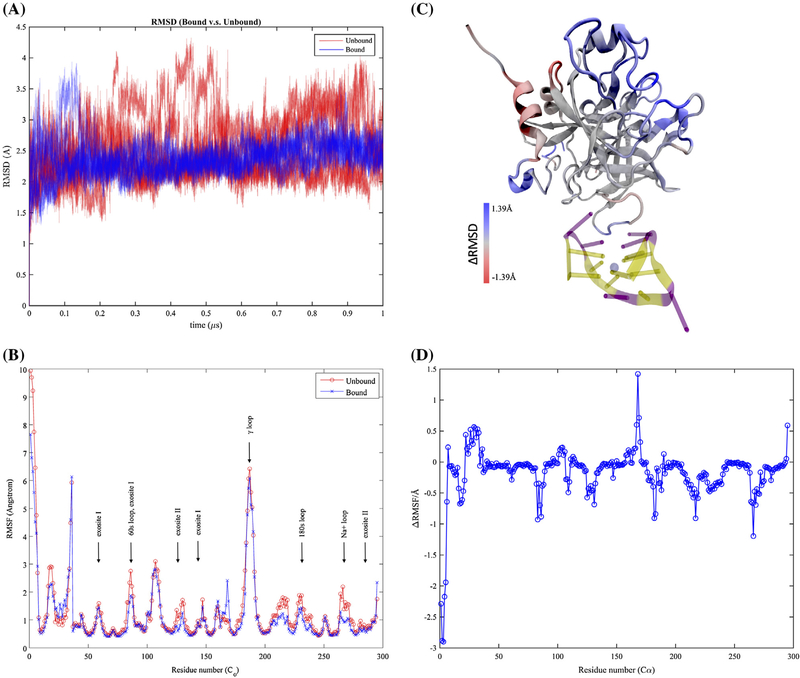
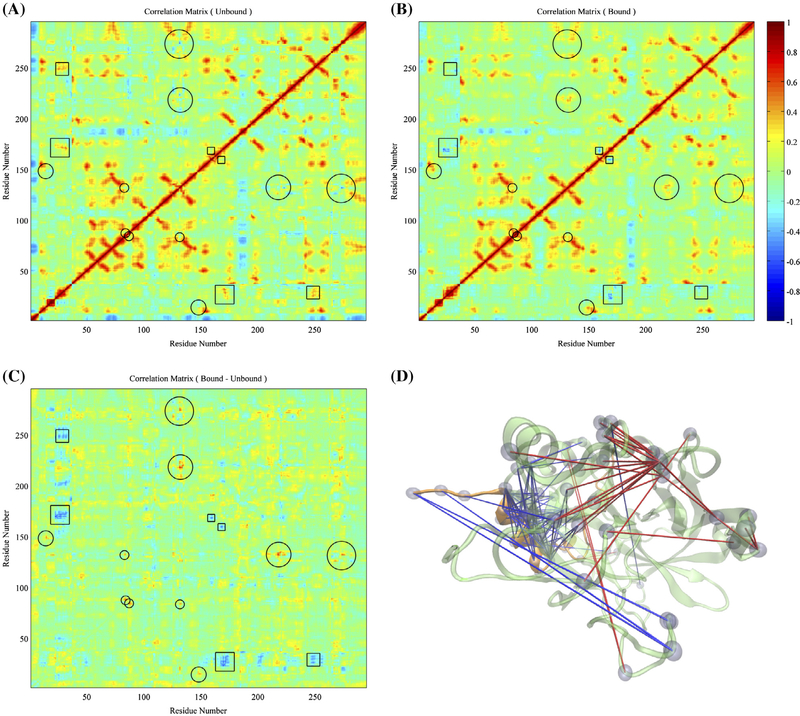
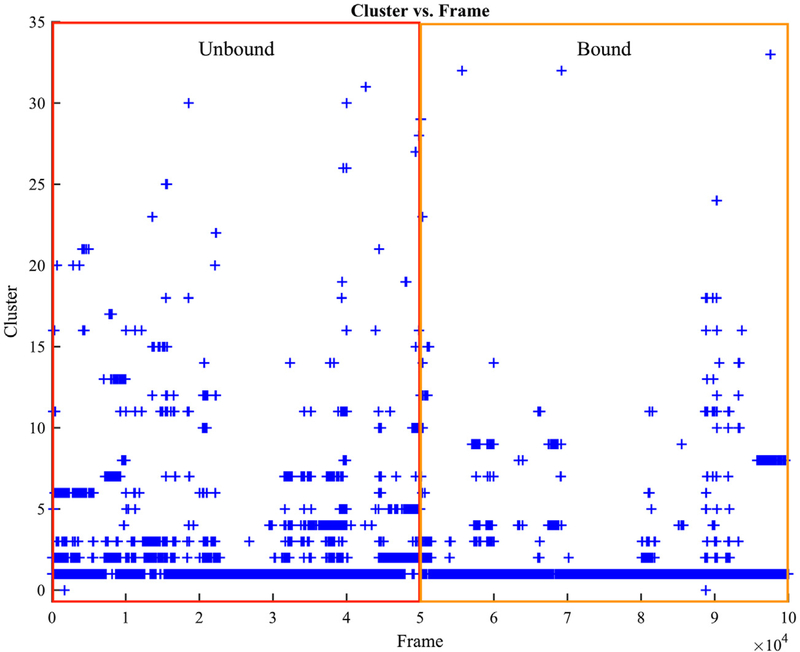
Thrombin is an attractive target for antithrombotic therapy due to its central role in thrombosis and hemostasis as well as its role in inducing tumor growth, metastasis, and tumor invasion. The thrombin-binding DNA aptamer (TBA), is under investigation for anticoagulant drugs. Although aptamer binding experiments have been revealed various effects on thrombin's enzymatic activities, the detailed picture of the thrombin's allostery from TBA binding is still unclear. To investigate thrombin's response to the aptamer-binding at the molecular level, we compare the mechanical properties and free energy landscapes of the free and aptamer-bound thrombin using microsecond-scale all-atom GPU-based molecular dynamics simulations. Our calculations on residue fluctuations and coupling illustrate the allosteric effects of aptamer-binding at the atomic level, highlighting the exosite II, 60s, γ and the sodium loops, and the alpha helix region in the light chains involved in the allosteric changes. This level of details clarifies the mechanisms of previous experimentally demonstrated phenomena, and provides a prediction of the reduced autolysis rate after aptamer-binding. The shifts in thrombin's ensemble of conformations and free energy surfaces after aptamer-binding demonstrate that the presence of bound-aptamer restricts the conformational freedom of thrombin suggesting that conformational selection, i.e. generalized allostery, is the dominant mechanism of thrombin-aptamer binding. The profound perturbation on thrombin's mechanical and thermodynamic properties due to the aptamer-binding, which was revealed comprehensively as a generalized allostery in this work, may be exploited in further drug discovery and development.
Keywords: aptamer; generalized allostery; molecular dynamics; molecular recognition; thrombin.
Conflict of interest statement
Disclosure statement
The authors of this manuscript declare no conflicts of interest.
Figures
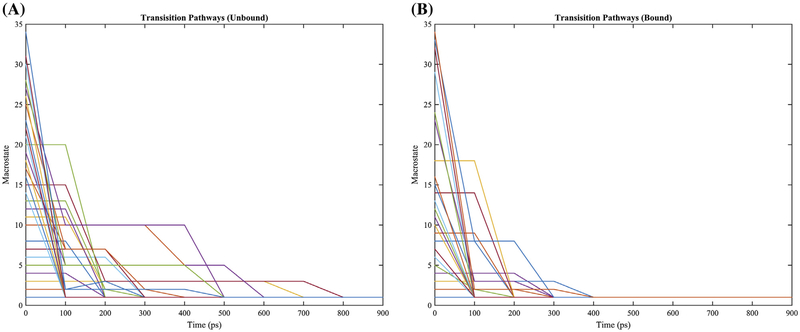
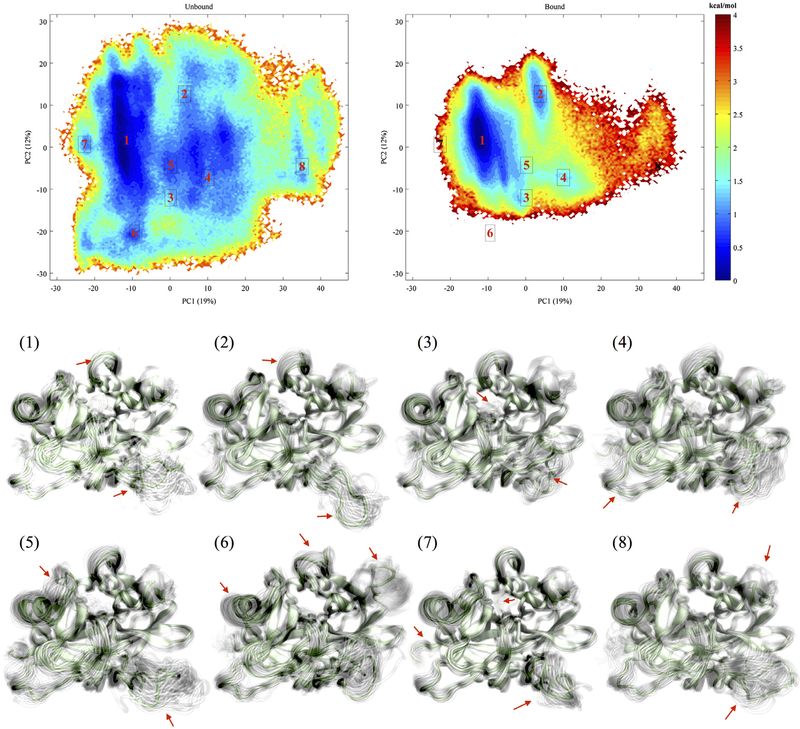
References
-
- Abdi H, & Williams LJ (2010). Principal component analysis. Wiley Interdisciplinary Reviews: Computational Statistics, 2, 433–459. doi: 10.1002/wics.101 - DOI
-
- Akhavan H, Mannucci PM, Lak H, Mancuso H, Mazzucconi MG, Rocino H, … Perkins SJ (2000). Identification and three-dimensional structural analysis of nine novel mutations in patients with prothrombin deficiency. Thrombosis and Haemostasis, 84, 989–997. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources