

Differential Infection Patterns and Recent Evolutionary Origins of Equine Hepaciviruses in Donkeys
- PMID: 27795428
- PMCID: PMC5165184
- DOI: 10.1128/JVI.01711-16
Differential Infection Patterns and Recent Evolutionary Origins of Equine Hepaciviruses in Donkeys
Abstract
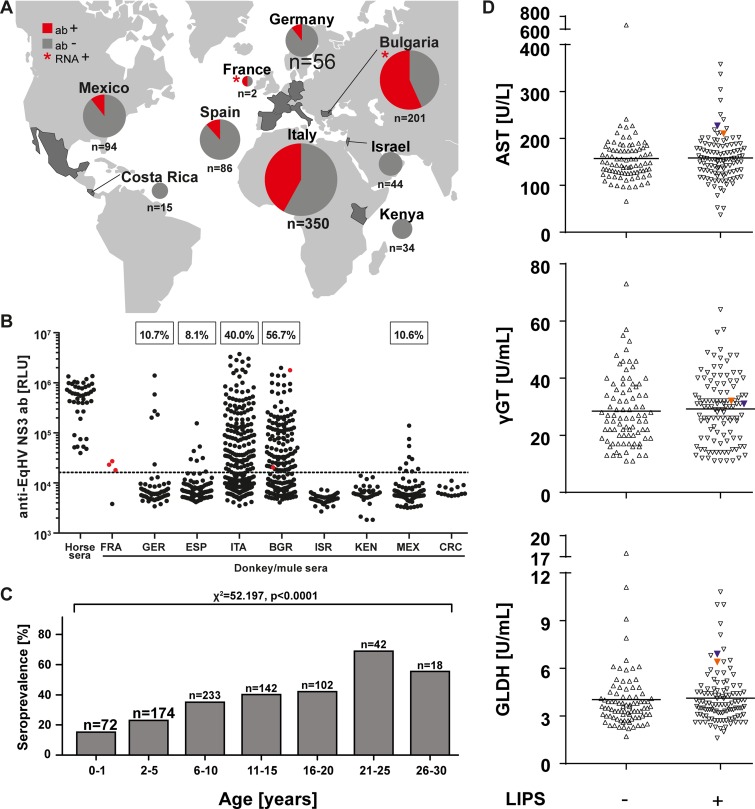
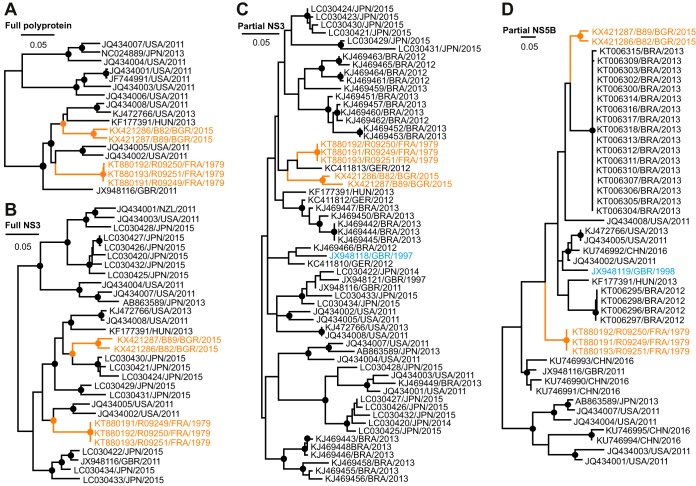
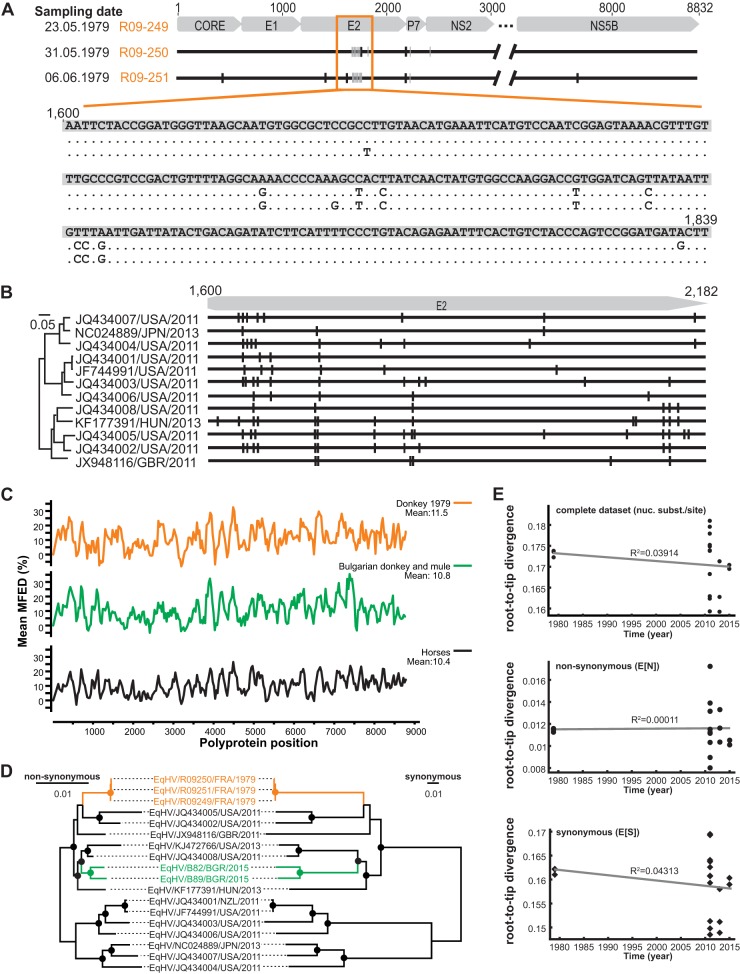
The hepatitis C virus (HCV) is a major human pathogen. Genetically related viruses in animals suggest a zoonotic origin of HCV. The closest relative of HCV is found in horses (termed equine hepacivirus [EqHV]). However, low EqHV genetic diversity implies relatively recent acquisition of EqHV by horses, making a derivation of HCV from EqHV unlikely. To unravel the EqHV evolutionary history within equid sister species, we analyzed 829 donkeys and 53 mules sampled in nine European, Asian, African, and American countries by molecular and serologic tools for EqHV infection. Antibodies were found in 278 animals (31.5%), and viral RNA was found in 3 animals (0.3%), all of which were simultaneously seropositive. A low RNA prevalence in spite of high seroprevalence suggests a predominance of acute infection, a possible difference from the mostly chronic hepacivirus infection pattern seen in horses and humans. Limitation of transmission due to short courses of infection may explain the existence of entirely seronegative groups of animals. Donkey and horse EqHV strains were paraphyletic and 97.5 to 98.2% identical in their translated polyprotein sequences, making virus/host cospeciation unlikely. Evolutionary reconstructions supported host switches of EqHV between horses and donkeys without the involvement of adaptive evolution. Global admixture of donkey and horse hepaciviruses was compatible with anthropogenic alterations of EqHV ecology. In summary, our findings do not support EqHV as the origin of the significantly more diversified HCV. Identification of a host system with predominantly acute hepacivirus infection may enable new insights into the chronic infection pattern associated with HCV.
Importance: The evolutionary origins of the human hepatitis C virus (HCV) are unclear. The closest animal-associated relative of HCV occurs in horses (equine hepacivirus [EqHV]). The low EqHV genetic diversity implies a relatively recent acquisition of EqHV by horses, limiting the time span for potential horse-to-human infections in the past. Horses are genetically related to donkeys, and EqHV may have cospeciated with these host species. Here, we investigated a large panel of donkeys from various countries using serologic and molecular tools. We found EqHV to be globally widespread in donkeys and identify potential differences in EqHV infection patterns, with donkeys potentially showing enhanced EqHV clearance compared to horses. We provide strong evidence against EqHV cospeciation and for its capability to switch hosts among equines. Differential hepacivirus infection patterns in horses and donkeys may enable new insights into the chronic infection pattern associated with HCV.
Keywords: donkey; equine hepacivirus; evolution; hepatitis C virus; pathogenesis.
Copyright © 2016 American Society for Microbiology.
Figures
References
-
- Global Burden of Disease Study. 2015. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386:743–800. doi: 10.1016/S0140-6736(15)60692-4. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
