

Carbon Monoxide Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by the Cyclic GMP/Protein Kinase G and NF-κB Signaling Pathway
- PMID: 27795439
- PMCID: PMC5165190
- DOI: 10.1128/JVI.01866-16
Carbon Monoxide Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by the Cyclic GMP/Protein Kinase G and NF-κB Signaling Pathway
Abstract
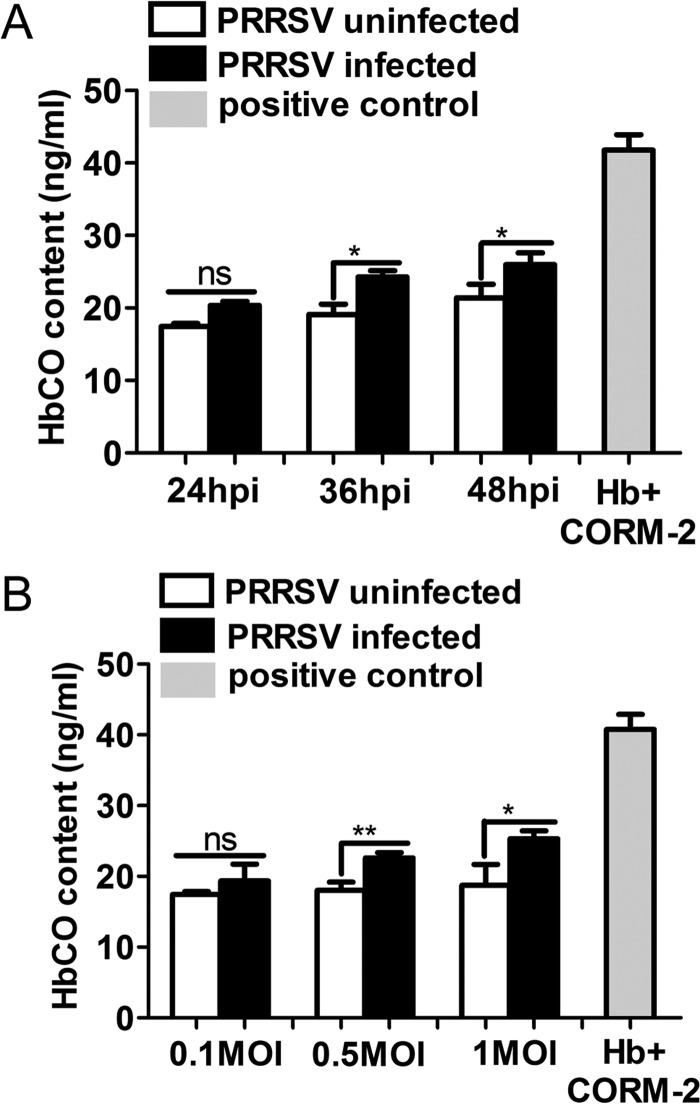
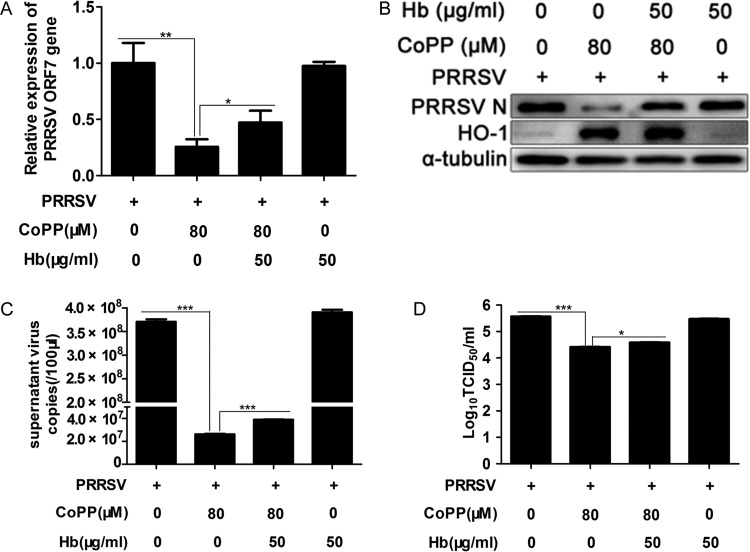
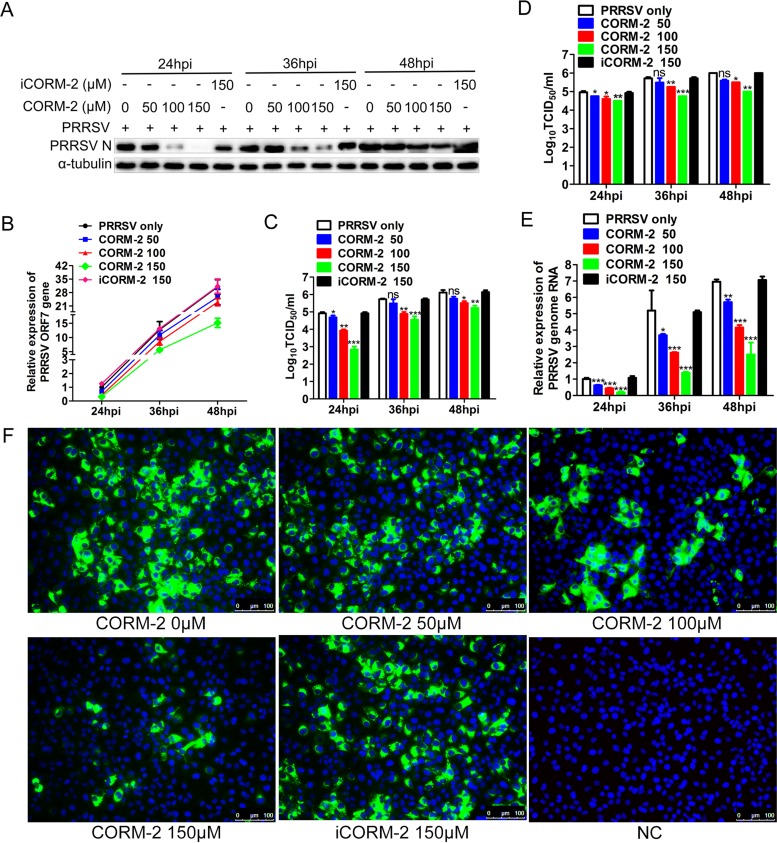
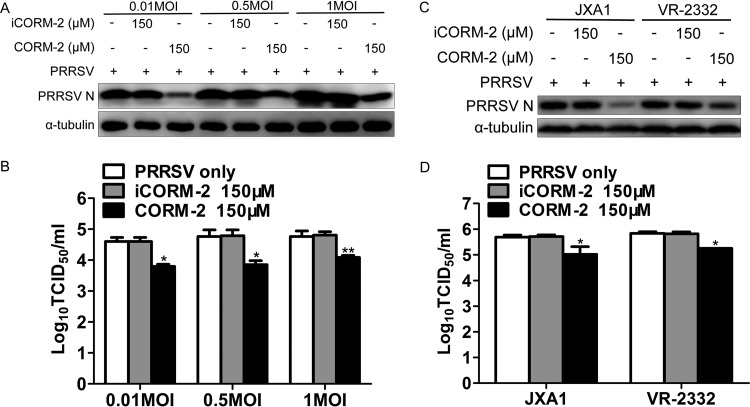
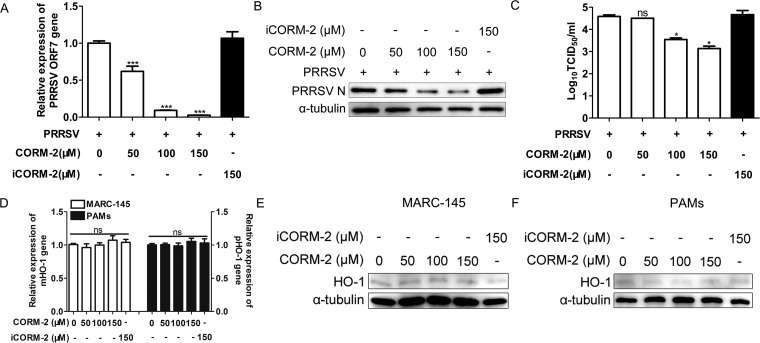
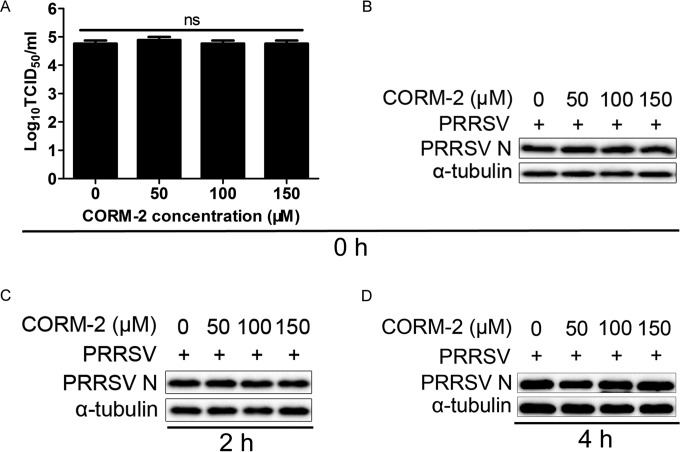
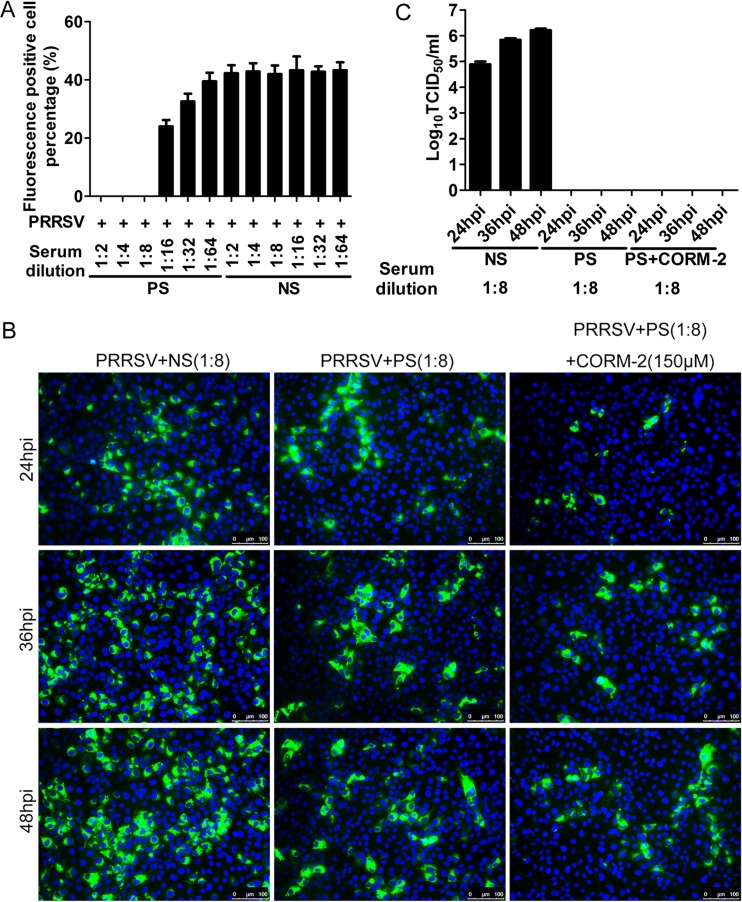
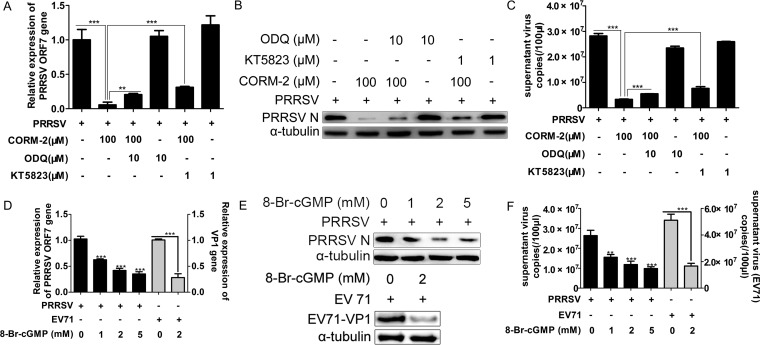
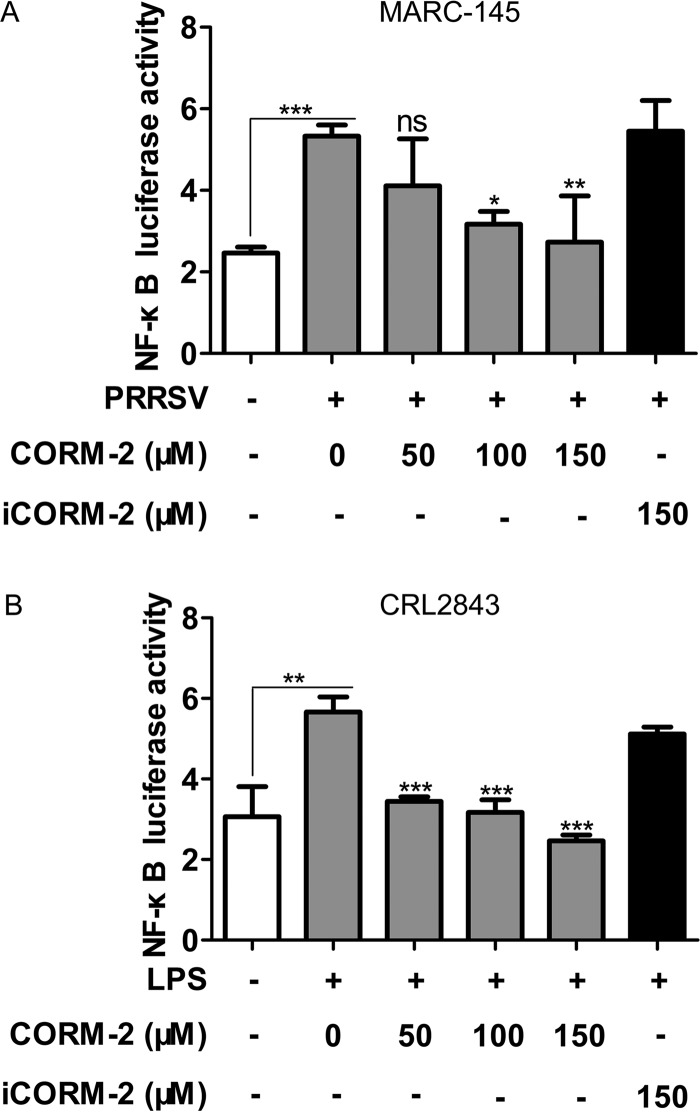
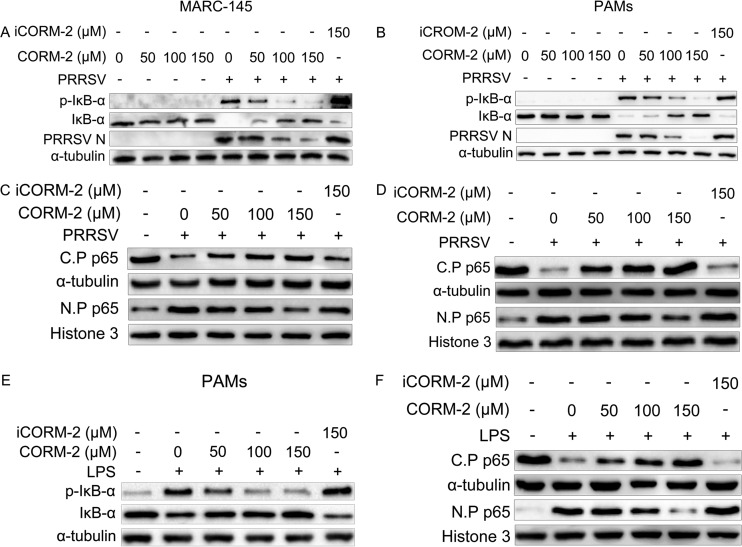
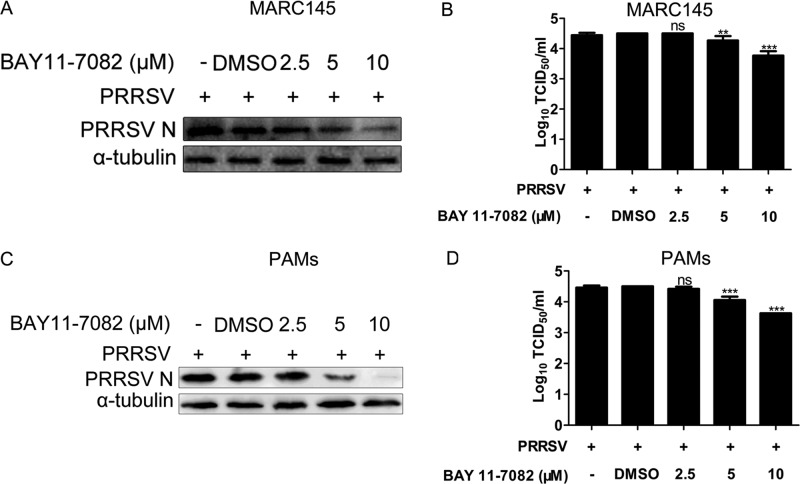
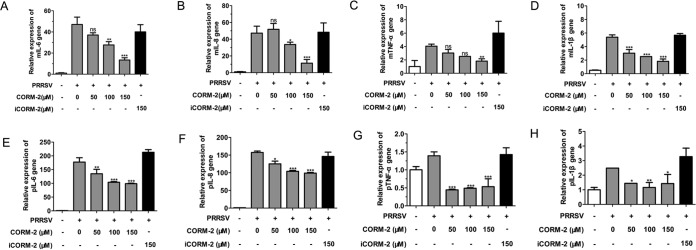
Porcine reproductive and respiratory syndrome virus (PRRSV) causes significant economic losses to the pork industry worldwide each year. Our previous research demonstrated that heme oxygenase-1 (HO-1) can suppress PRRSV replication via an unknown molecular mechanism. In this study, inhibition of PRRSV replication was demonstrated to be mediated by carbon monoxide (CO), a downstream metabolite of HO-1. Using several approaches, we demonstrate that CO significantly inhibited PRRSV replication in both a PRRSV permissive cell line, MARC-145, and the predominant cell type targeted during in vivo PRRSV infection, porcine alveolar macrophages (PAMs). Our results showed that CO inhibited intercellular spread of PRRSV; however, it did not affect PRRSV entry into host cells. Furthermore, CO was found to suppress PRRSV replication via the activation of the cyclic GMP/protein kinase G (cGMP/PKG) signaling pathway. CO significantly inhibits PRRSV-induced NF-κB activation, a required step for PRRSV replication. Moreover, CO significantly reduced PRRSV-induced proinflammatory cytokine mRNA levels. In conclusion, the present study demonstrates that CO exerts its anti-PRRSV effect by activating the cellular cGMP/PKG signaling pathway and by negatively regulating cellular NF-κB signaling. These findings not only provide new insights into the molecular mechanism of HO-1 inhibition of PRRSV replication but also suggest potential new control measures for future PRRSV outbreaks.
Importance: PRRSV causes great economic losses each year to the swine industry worldwide. Carbon monoxide (CO), a metabolite of HO-1, has been shown to have antimicrobial and antiviral activities in infected cells. Our previous research demonstrated that HO-1 can suppress PRRSV replication. Here we show that endogenous CO produced through HO-1 catalysis mediates the antiviral effect of HO-1. CO inhibits PRRSV replication by activating the cellular cGMP/PKG signaling pathway and by negatively regulating cellular NF-κB signaling. These findings not only provide new insights into the molecular mechanism of HO-1 inhibition of PRRSV replication but also suggest potential new control measures for future PRRSV outbreaks.
Keywords: HO-1; NF-κB; PRRSV; cGMP/PKG; carbon monoxide.
Copyright © 2016 American Society for Microbiology.
Figures
References
-
- Cavanagh D. 1997. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol 142:629–633. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
