

Cox-2 Inhibition Protects against Hypoxia/Reoxygenation-Induced Cardiomyocyte Apoptosis via Akt-Dependent Enhancement of iNOS Expression
- PMID: 27795807
- PMCID: PMC5067333
- DOI: 10.1155/2016/3453059
Cox-2 Inhibition Protects against Hypoxia/Reoxygenation-Induced Cardiomyocyte Apoptosis via Akt-Dependent Enhancement of iNOS Expression
Abstract
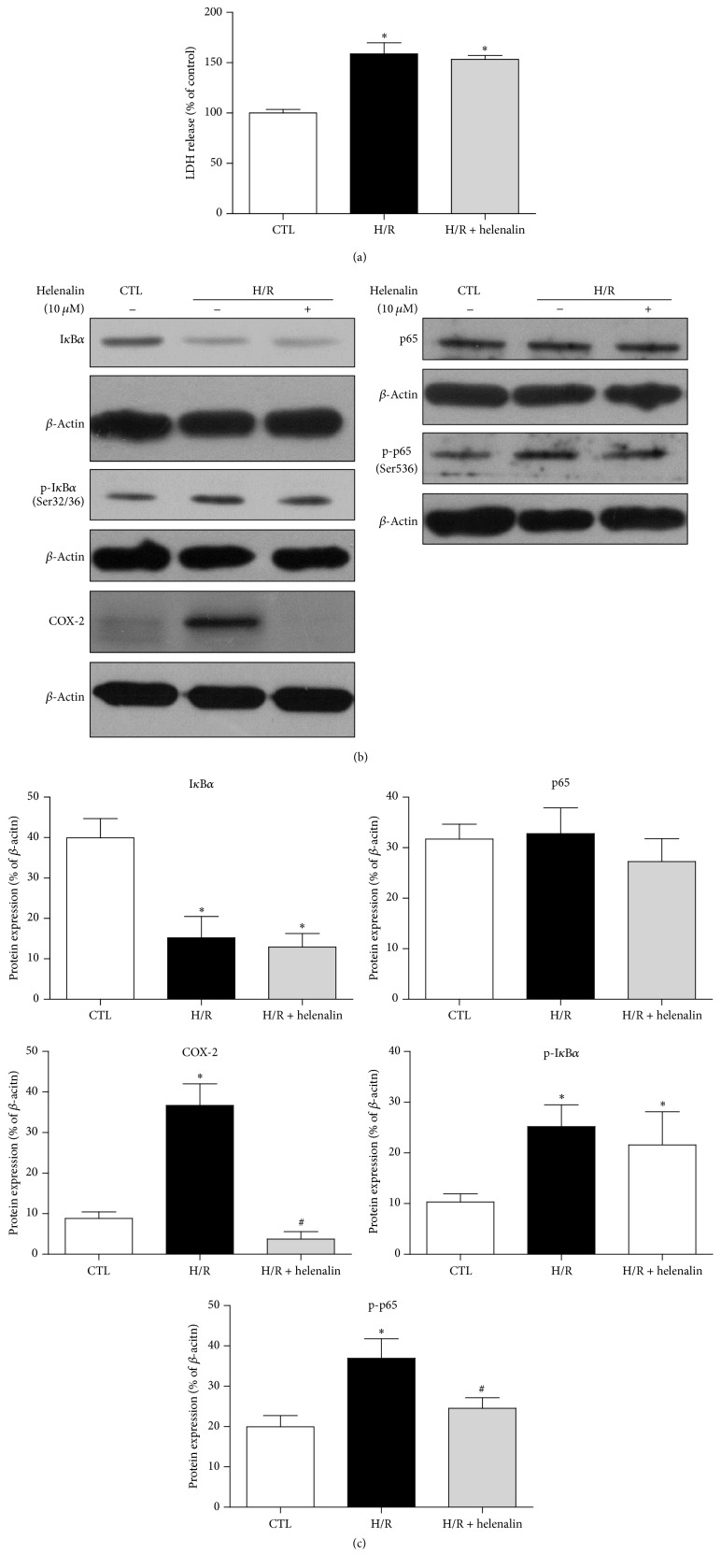
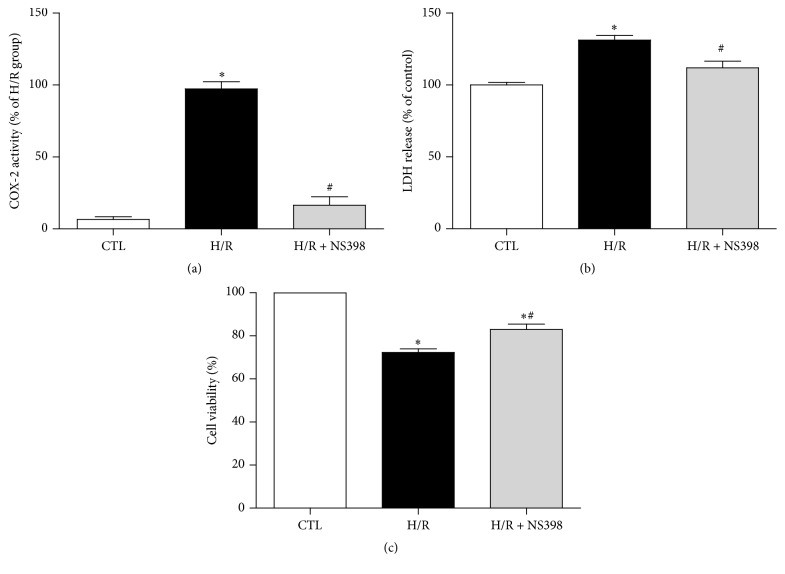
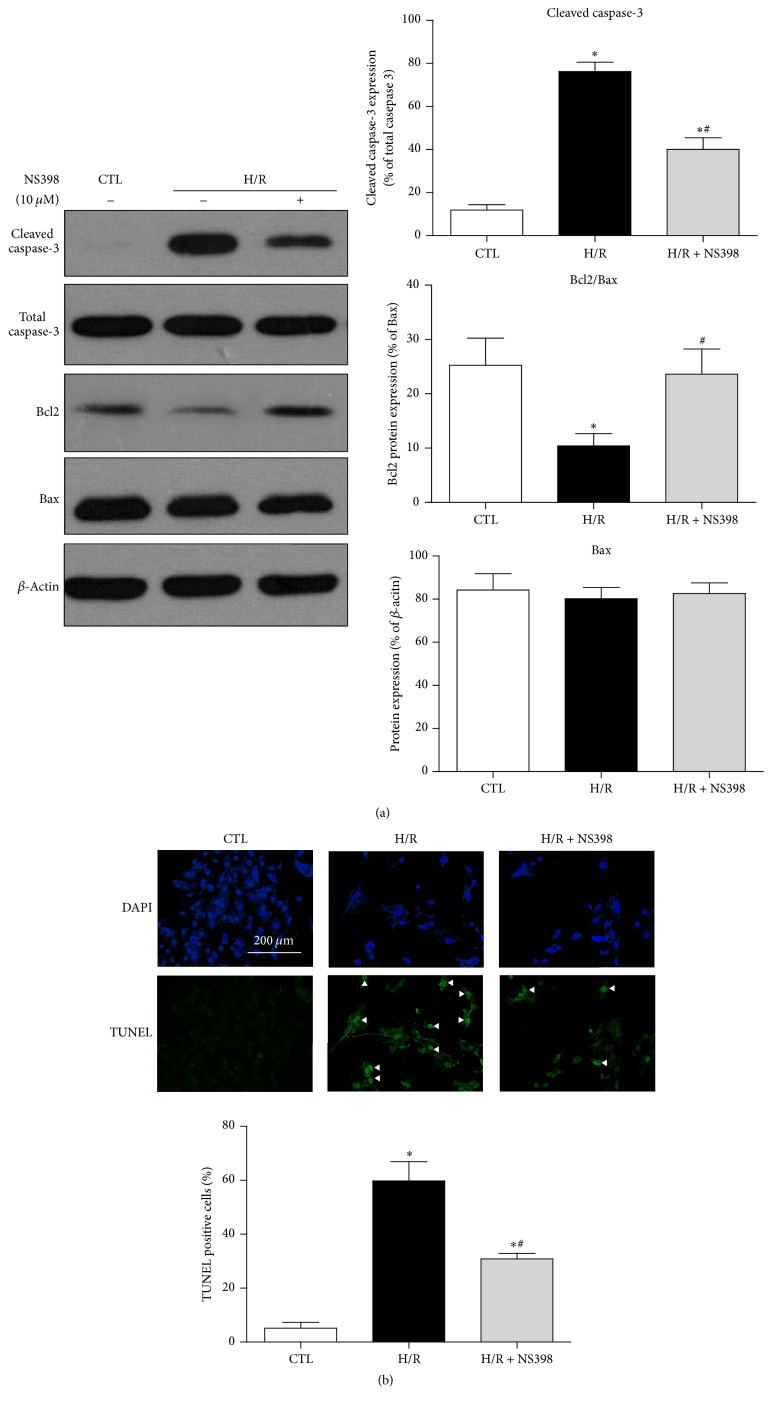
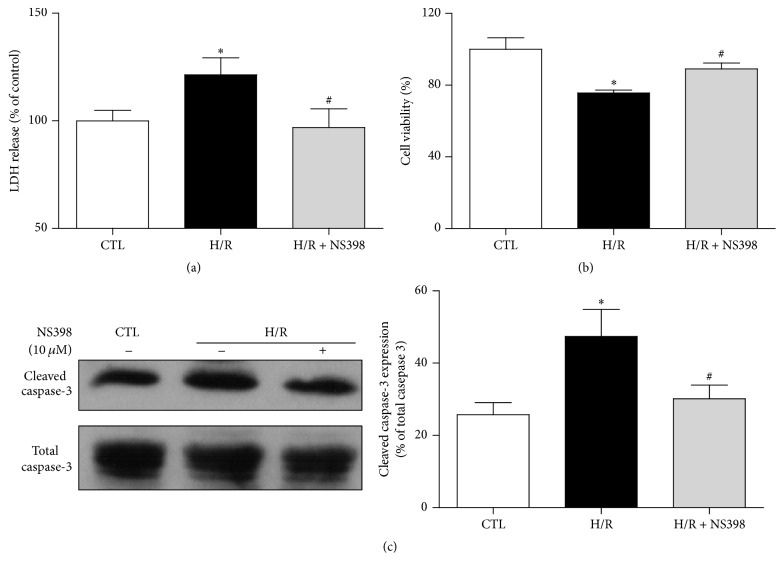
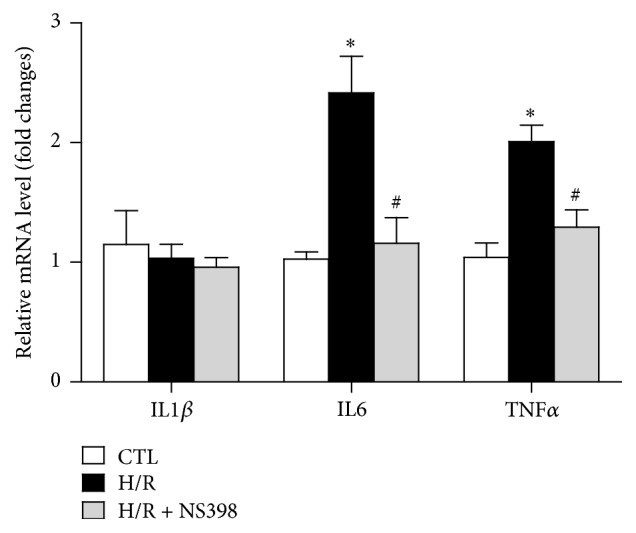
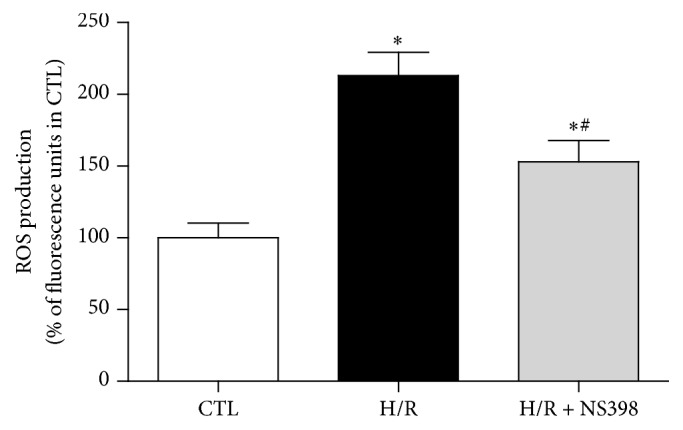
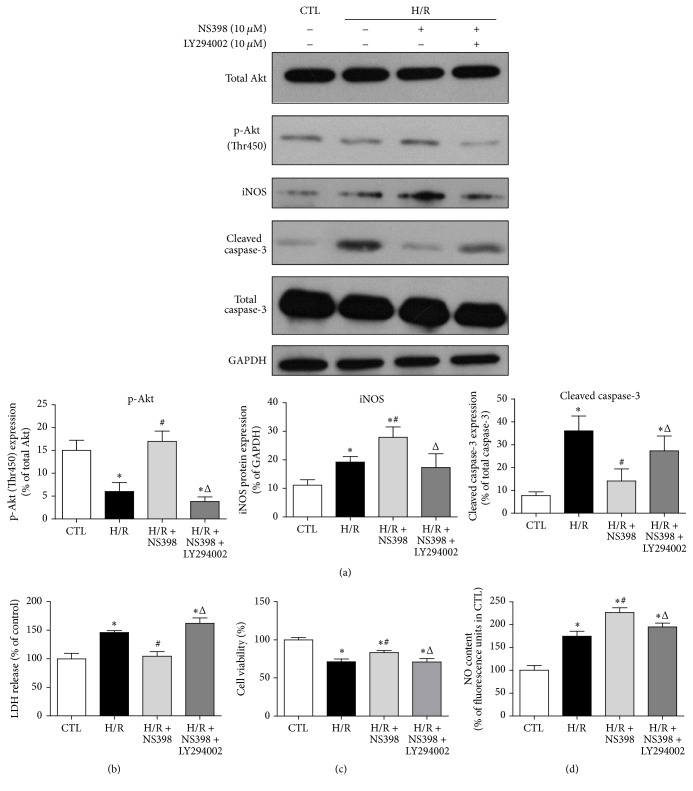
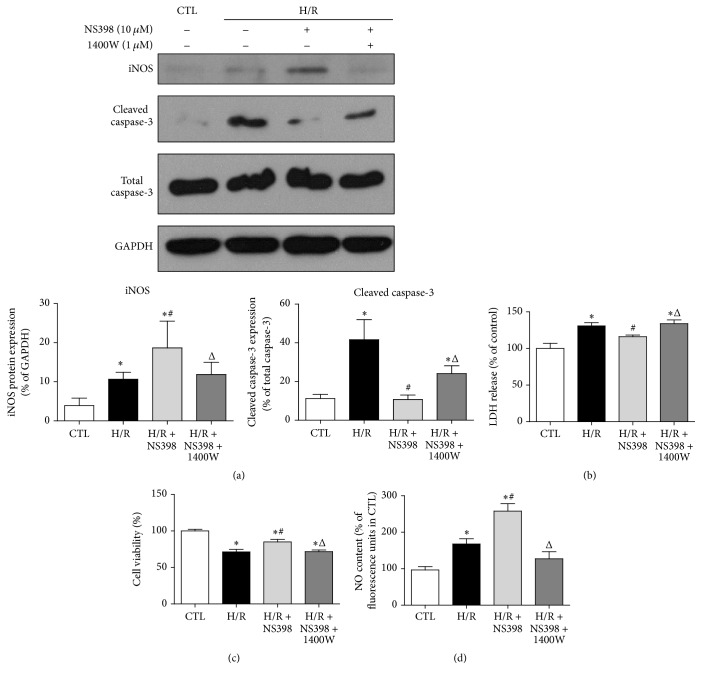
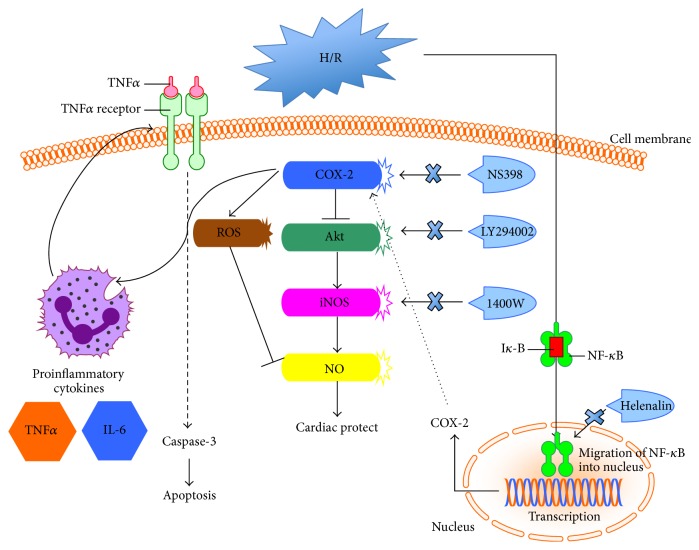
The present study explored the potential causal link between ischemia-driven cyclooxygenase-2 (COX-2) expression and enhanced apoptosis during myocardial ischemia/reperfusion (I/R) by using H9C2 cardiomyocytes and primary rat cardiomyocytes subjected to hypoxia/reoxygenation (H/R). The results showed that H/R resulted in higher COX-2 expression than that of controls, which was prevented by pretreatment with Helenalin (NFκB specific inhibitor). Furthermore, pretreatment with NS398 (COX-2 specific inhibitor) significantly attenuated H/R-induced cell injury [lower lactate dehydrogenase (LDH) leakage and enhanced cell viability] and apoptosis (higher Bcl2 expression and lower level of cleaved caspases-3 and TUNEL-positive cells) in cardiomyocytes. The amelioration of posthypoxic apoptotic cell death was paralleled by significant attenuation of H/R-induced increases in proinflammatory cytokines [interleukin 6 (IL6) and tumor necrosis factor (TNFα)] and reactive oxygen species (ROS) production and by higher protein expression of phosphorylated Akt and inducible nitric oxide synthase (iNOS) and enhanced nitric oxide production. Moreover, the application of LY294002 (Akt-specific inhibitor) or 1400W (iNOS-selective inhibitor) cancelled the cellular protective effects of NS398. Findings from the current study suggest that activation of NFκB during cardiomyocyte H/R induces the expression of COX-2 and that higher COX-2 expression during H/R exacerbates cardiomyocyte H/R injury via mechanisms that involve cross talks among inflammation, ROS, and Akt/iNOS/NO signaling.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
