

Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis
- PMID: 27798841
- PMCID: PMC5088563
- DOI: 10.1101/gad.289298.116
Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis
Abstract
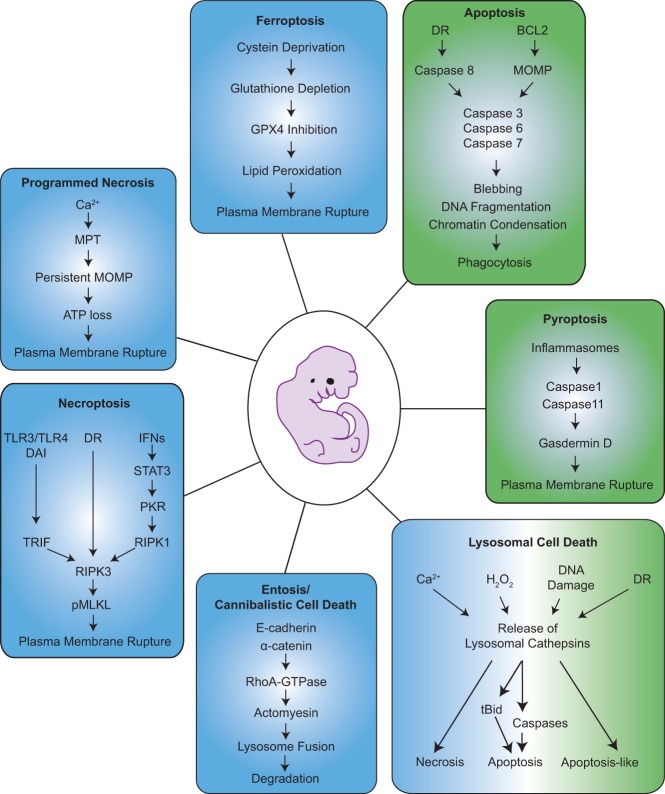
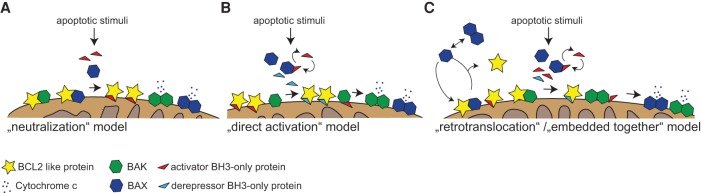
"Programmed cell death or 'apoptosis' is critical for organogenesis during embryonic development and tissue homeostasis in the adult. Its deregulation can contribute to a broad range of human pathologies, including neurodegeneration, cancer, or autoimmunity…" These or similar phrases have become generic opening statements in many reviews and textbooks describing the physiological relevance of apoptotic cell death. However, while the role in disease has been documented beyond doubt, facilitating innovative drug discovery, we wonder whether the former is really true. What goes wrong in vertebrate development or in adult tissue when the main route to apoptotic cell death, controlled by the BCL2 family, is impaired? Such scenarios have been mimicked by deletion of one or more prodeath genes within the BCL2 family, and gene targeting studies in mice exploring the consequences have been manifold. Many of these studies were geared toward understanding the role of BCL2 family proteins and mitochondrial apoptosis in disease, whereas fewer focused in detail on their role during normal development or tissue homeostasis, perhaps also due to an irritating lack of phenotype. Looking at these studies, the relevance of classical programmed cell death by apoptosis for development appears rather limited. Together, these many studies suggest either highly selective and context-dependent contributions of mitochondrial apoptosis or significant redundancy with alternative cell death mechanisms, as summarized and discussed here.
Keywords: BCL2 family; BH3-only proteins; cell death; development; tissue homeostasis.
© 2016 Tuzlak et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Apoptosis: a mitochondrial perspective on cell death.Indian J Exp Biol. 2005 Jan;43(1):25-34. Indian J Exp Biol. 2005. PMID: 15691062 Review.
-
Bcl2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway.Hum Mol Genet. 2001 Oct 1;10(21):2329-39. doi: 10.1093/hmg/10.21.2329. Hum Mol Genet. 2001. PMID: 11689480
-
Interaction of E1B 19K with Bax is required to block Bax-induced loss of mitochondrial membrane potential and apoptosis.Oncogene. 1998 Dec 10;17(23):2993-3005. doi: 10.1038/sj.onc.1202215. Oncogene. 1998. PMID: 9881701
-
BH3-only proteins and their roles in programmed cell death.Oncogene. 2008 Dec;27 Suppl 1:S128-36. doi: 10.1038/onc.2009.50. Oncogene. 2008. PMID: 19641498 Review.
-
Homeostatic functions of BCL-2 proteins beyond apoptosis.Adv Exp Med Biol. 2010;687:1-32. doi: 10.1007/978-1-4419-6706-0_1. Adv Exp Med Biol. 2010. PMID: 20919635 Review.
Cited by
-
Apoptosis as a Barrier against CIN and Aneuploidy.Cancers (Basel). 2022 Dec 21;15(1):30. doi: 10.3390/cancers15010030. Cancers (Basel). 2022. PMID: 36612027 Free PMC article. Review.
-
p62/SQSTM1-induced caspase-8 aggresomes are essential for ionizing radiation-mediated apoptosis.Cell Death Dis. 2021 Oct 25;12(11):997. doi: 10.1038/s41419-021-04301-7. Cell Death Dis. 2021. PMID: 34697296 Free PMC article.
-
Apoptosis-50 Years after Its Discovery.Biomedicines. 2023 Apr 18;11(4):1196. doi: 10.3390/biomedicines11041196. Biomedicines. 2023. PMID: 37189814 Free PMC article.
-
From Cell Death to Regeneration: Rebuilding After Injury.Front Cell Dev Biol. 2021 Mar 18;9:655048. doi: 10.3389/fcell.2021.655048. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33816506 Free PMC article. Review.
-
Checkpoint kinase 1 is essential for fetal and adult hematopoiesis.EMBO Rep. 2019 Aug;20(8):e47026. doi: 10.15252/embr.201847026. Epub 2019 Jun 17. EMBO Rep. 2019. PMID: 31379128 Free PMC article.
References
-
- Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, Lebois M, Hakem R, Josefsson EC, O'Reilly LA, et al. 2016. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45: 513–526. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources