

Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations
- PMID: 27799555
- PMCID: PMC5135377
- DOI: 10.1073/pnas.1603754113
Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations
Abstract
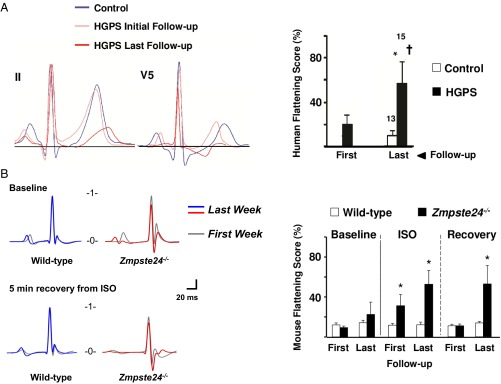
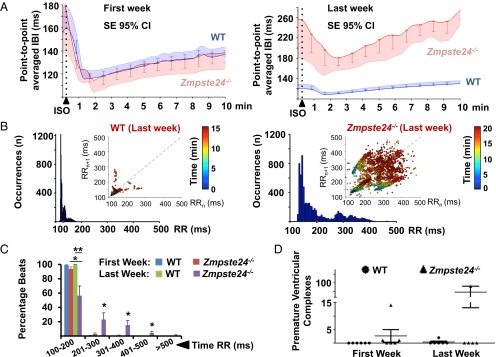
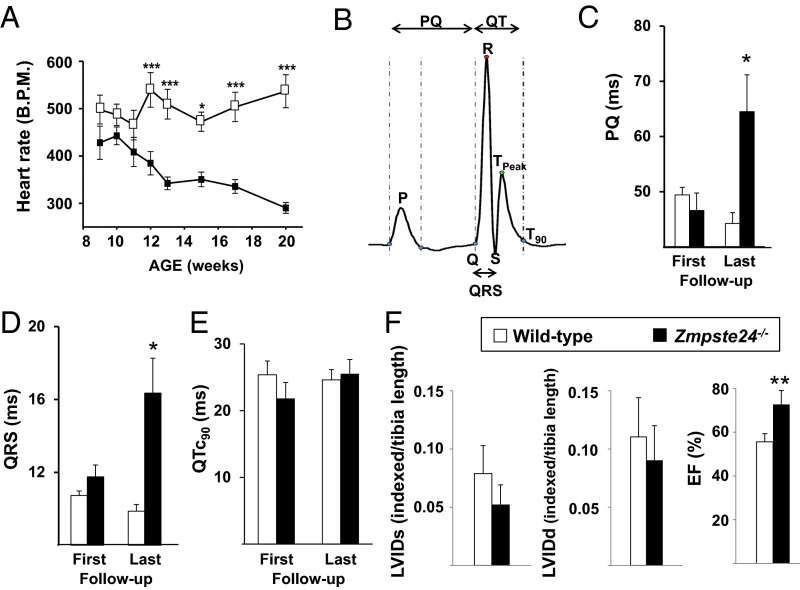
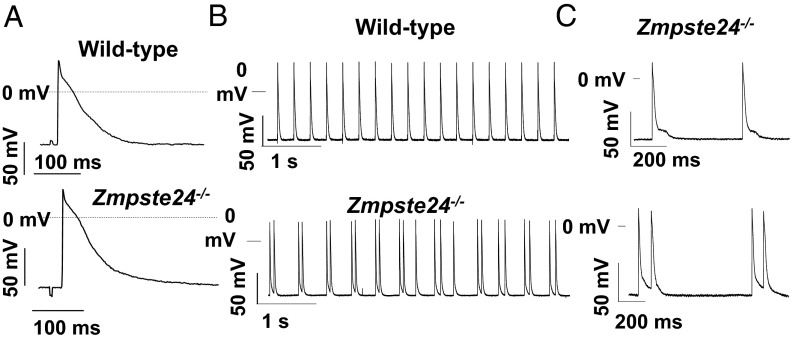
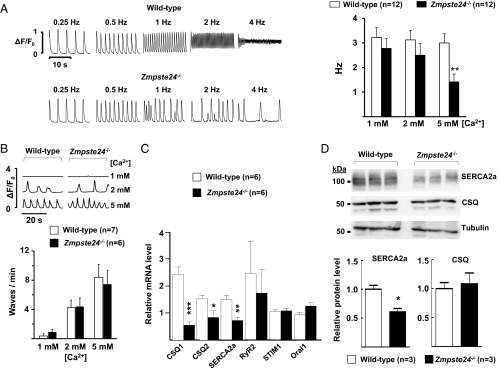
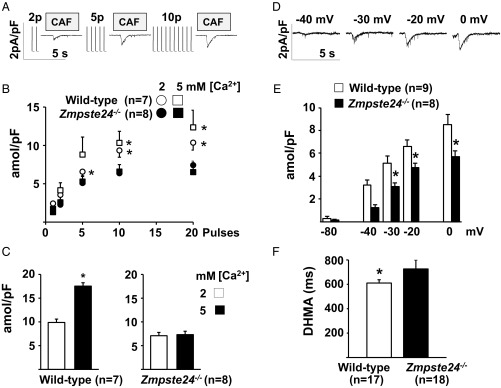
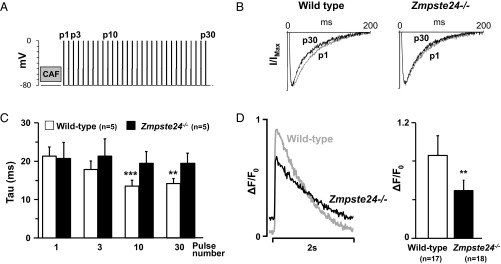
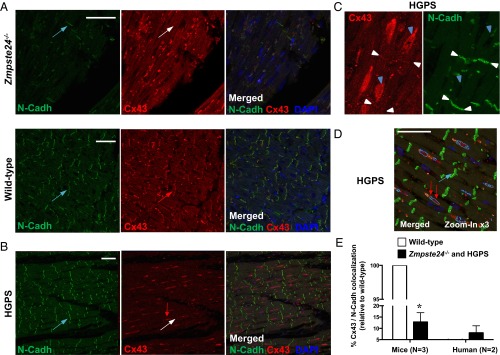
Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disease caused by defective prelamin A processing, leading to nuclear lamina alterations, severe cardiovascular pathology, and premature death. Prelamin A alterations also occur in physiological aging. It remains unknown how defective prelamin A processing affects the cardiac rhythm. We show age-dependent cardiac repolarization abnormalities in HGPS patients that are also present in the Zmpste24-/- mouse model of HGPS. Challenge of Zmpste24-/- mice with the β-adrenergic agonist isoproterenol did not trigger ventricular arrhythmia but caused bradycardia-related premature ventricular complexes and slow-rate polymorphic ventricular rhythms during recovery. Patch-clamping in Zmpste24-/- cardiomyocytes revealed prolonged calcium-transient duration and reduced sarcoplasmic reticulum calcium loading and release, consistent with the absence of isoproterenol-induced ventricular arrhythmia. Zmpste24-/- progeroid mice also developed severe fibrosis-unrelated bradycardia and PQ interval and QRS complex prolongation. These conduction defects were accompanied by overt mislocalization of the gap junction protein connexin43 (Cx43). Remarkably, Cx43 mislocalization was also evident in autopsied left ventricle tissue from HGPS patients, suggesting intercellular connectivity alterations at late stages of the disease. The similarities between HGPS patients and progeroid mice reported here strongly suggest that defective cardiac repolarization and cardiomyocyte connectivity are important abnormalities in the HGPS pathogenesis that increase the risk of arrhythmia and premature death.
Keywords: Hutchinson–Gilford progeria syndrome; calcium handling; connexin43; prelamin A; progerin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Trigueros-Motos L, González JM, Rivera J, Andrés V. Hutchinson-Gilford progeria syndrome, cardiovascular disease and oxidative stress. Front Biosci (Schol Ed) 2011;3:1285–1297. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
