

Pulmonary fibrosis in the era of stratified medicine
- PMID: 27799632
- PMCID: PMC5450041
- DOI: 10.1136/thoraxjnl-2016-209172
Pulmonary fibrosis in the era of stratified medicine
Abstract
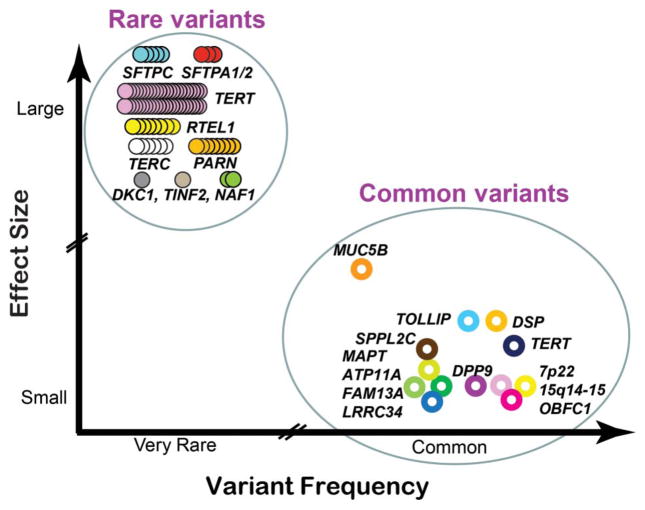
Both common and rare variants contribute to the genetic architecture of pulmonary fibrosis. Genome-wide association studies have identified common variants, or those with a minor allele frequency of >5%, that are linked to pulmonary fibrosis. The most widely replicated variant (rs35705950) is located in the promoter region of the MUC5B gene and has been strongly associated with idiopathic pulmonary fibrosis (IPF) and familial interstitial pneumonia (FIP) across multiple different cohorts. However, many more common variants have been identified with disease risk and in aggregate account for approximately one-third of the risk of IPF. Moreover, several of these common variants appear to have prognostic potential. Next generation sequencing technologies have facilitated the identification of rare variants. Recent whole exome sequencing studies have linked pathogenic rare variants in multiple new genes to FIP. Compared with common variants, rare variants have lower population allele frequencies and higher effect sizes. Pulmonary fibrosis rare variants genes can be subdivided into two pathways: telomere maintenance and surfactant metabolism. Heterozygous rare variants in telomere-related genes co-segregate with adult-onset pulmonary fibrosis with incomplete penetrance, lead to reduced protein function, and are associated with short telomere lengths. Despite poor genotype-phenotype correlations, lung fibrosis associated with pathogenic rare variants in different telomere genes is progressive and displays similar survival characteristics. In contrast, many of the heterozygous rare variants in the surfactant genes predict a gain of toxic function from protein misfolding and increased endoplasmic reticulum (ER) stress. Evidence of both telomere shortening and increased ER stress have been found in sporadic IPF patients, suggesting that the mechanisms identified from rare variant genetic studies in unique individuals and families are applicable to a wider spectrum of patients. The ability to sequence large cohorts of individuals rapidly has the potential to further our understanding of the relative contributions of common and rare variants in the pathogenesis of pulmonary fibrosis. The UK 100,000 Genomes Project will provide opportunities to interrogate both common and rare variants and to investigate how these biological signals provide diagnostic and prognostic information in the era of stratified medicine.
Keywords: Idiopathic pulmonary fibrosis; Interstitial Fibrosis.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/.
Conflict of interest statement
Competing interests: The authors have in the past received and currently receive funding from the National Institutes of Health.
Figures
References
-
- Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–84. - PubMed
-
- Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82. - PubMed
-
- King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical