

Loss of SynDIG1 Reduces Excitatory Synapse Maturation But Not Formation In Vivo
- PMID: 27800545
- PMCID: PMC5073248
- DOI: 10.1523/ENEURO.0130-16.2016
Loss of SynDIG1 Reduces Excitatory Synapse Maturation But Not Formation In Vivo
Abstract
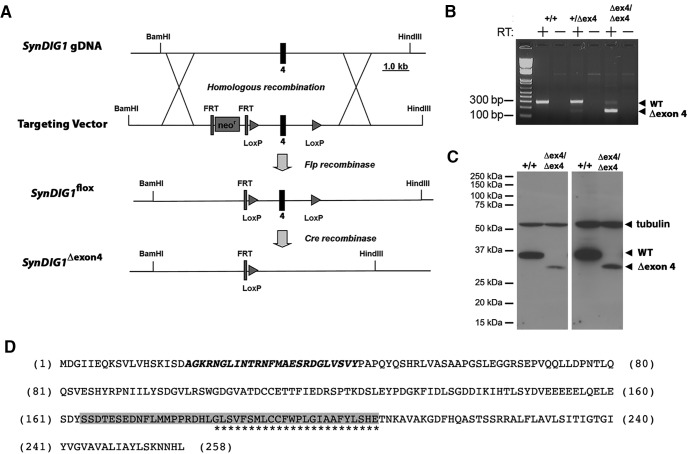
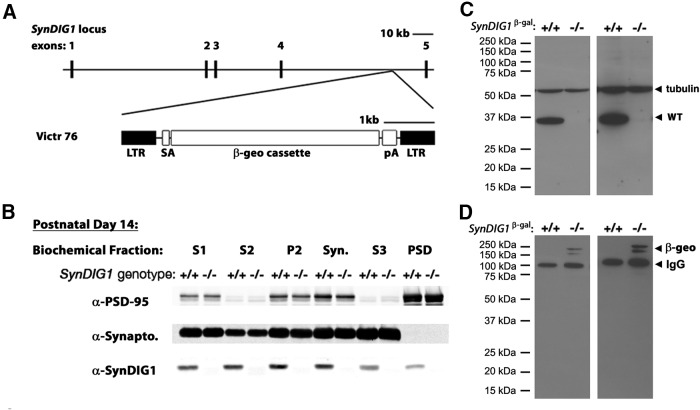
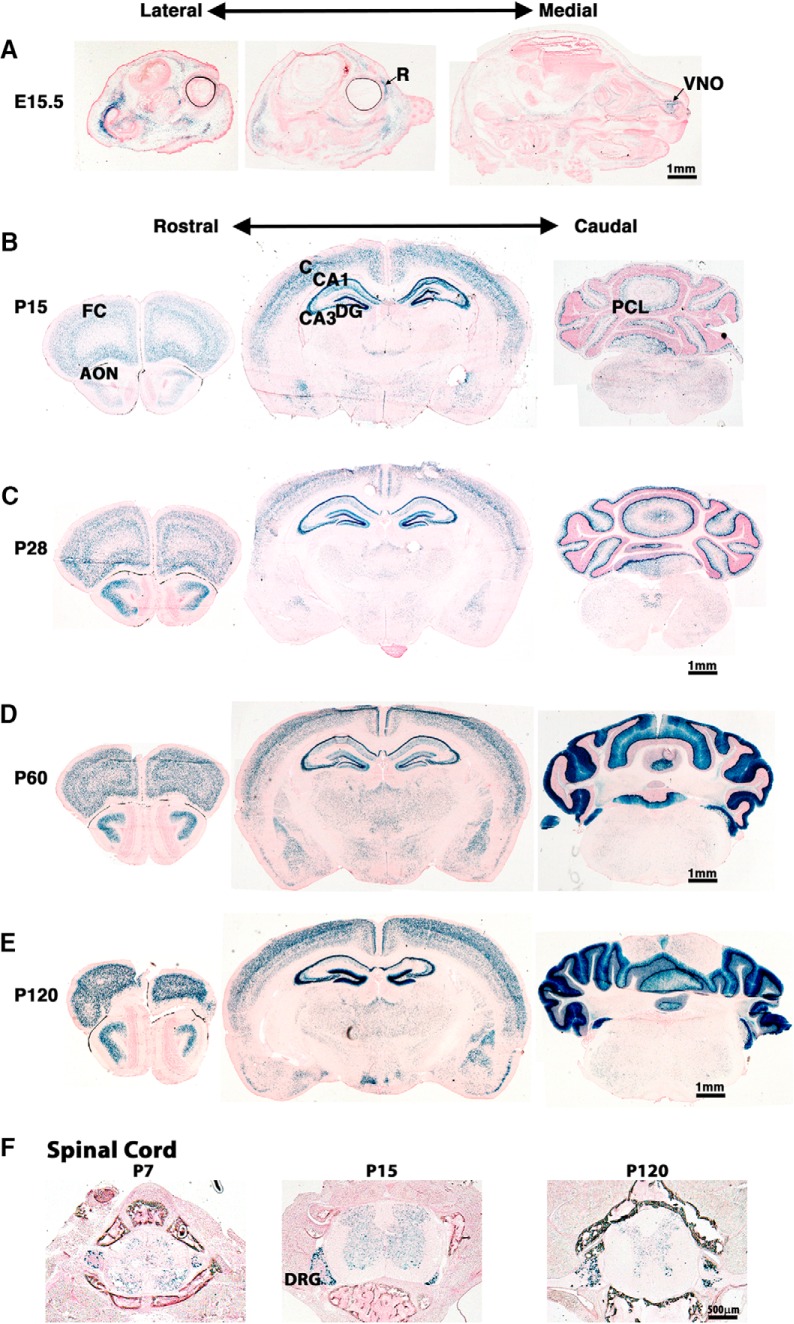
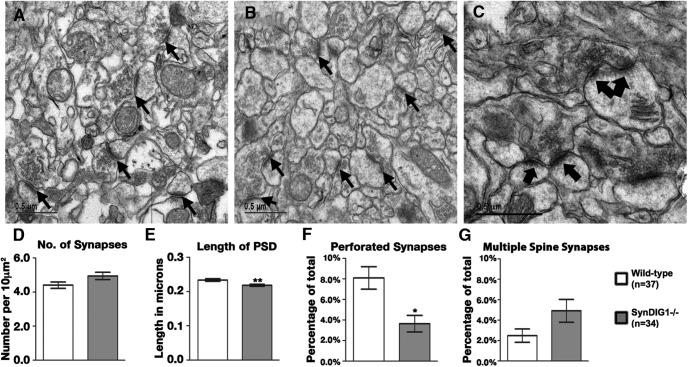
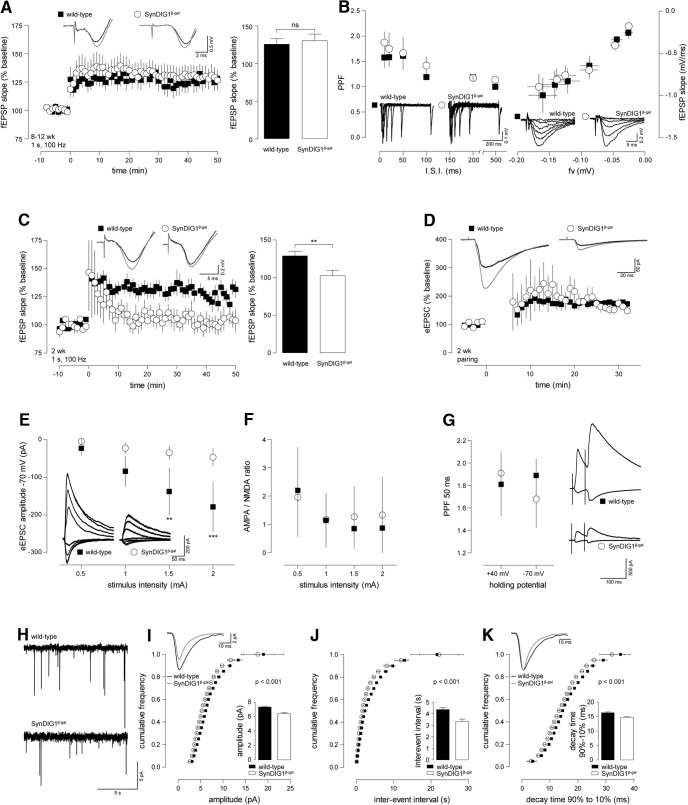
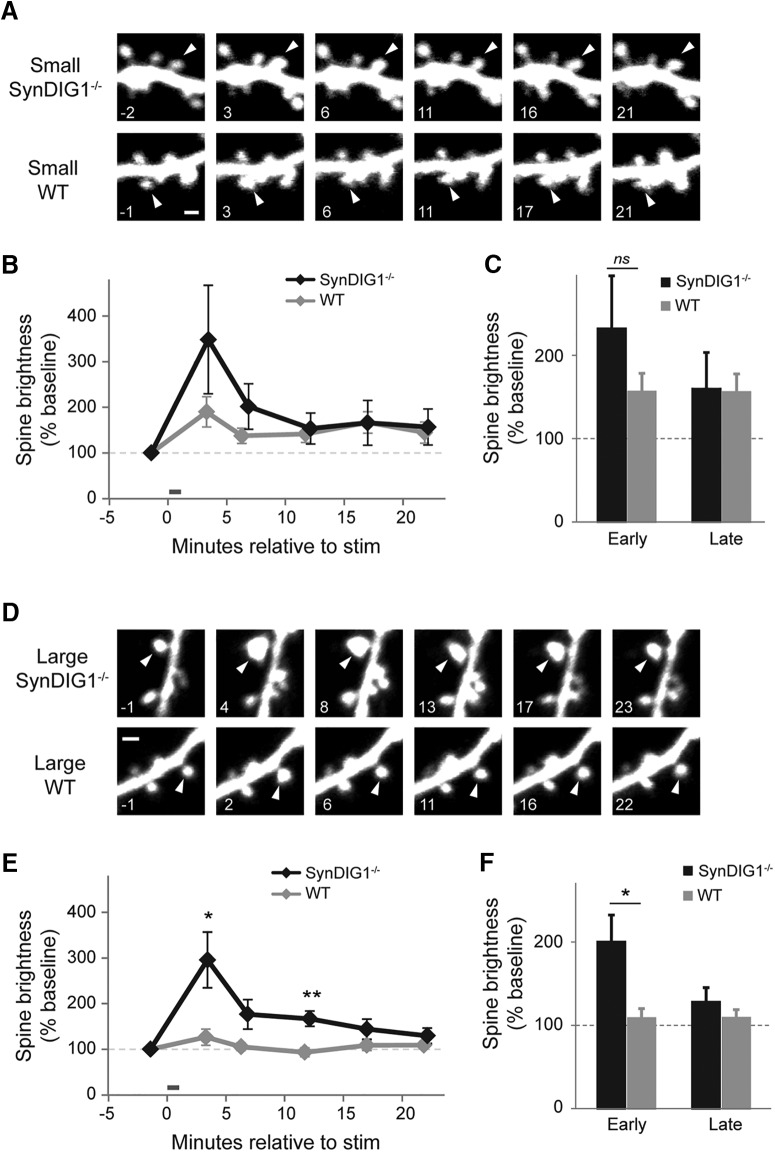
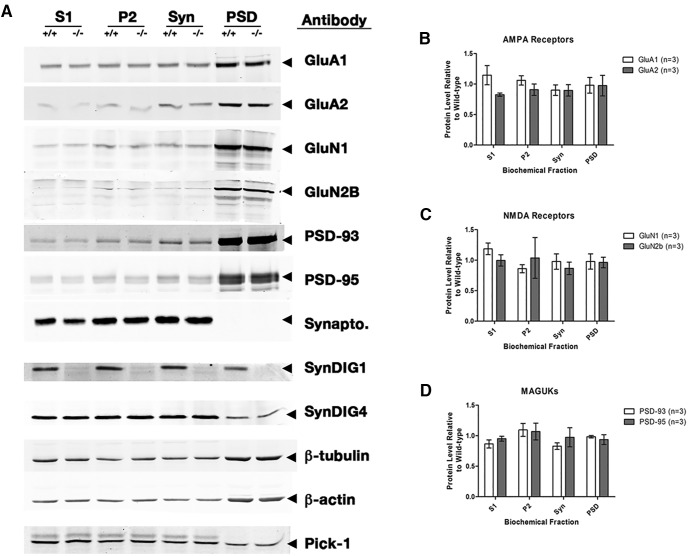
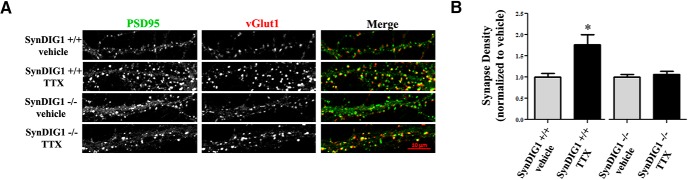
Modification of the strength of excitatory synaptic connections is a fundamental mechanism by which neural circuits are refined during development and learning. Synapse Differentiation Induced Gene 1 (SynDIG1) has been shown to play a key role in regulating synaptic strength in vitro. Here, we investigated the role of SynDIG1 in vivo in mice with a disruption of the SynDIG1 gene rather than use an alternate loxP-flanked conditional mutant that we find retains a partial protein product. The gene-trap insertion with a reporter cassette mutant mice shows that the SynDIG1 promoter is active during embryogenesis in the retina with some activity in the brain, and postnatally in the mouse hippocampus, cortex, hindbrain, and spinal cord. Ultrastructural analysis of the hippocampal CA1 region shows a decrease in the average PSD length of synapses and a decrease in the number of synapses with a mature phenotype. Intriguingly, the total synapse number appears to be increased in SynDIG1 mutant mice. Electrophysiological analyses show a decrease in AMPA and NMDA receptor function in SynDIG1-deficient hippocampal neurons. Glutamate stimulation of individual dendritic spines in hippocampal slices from SynDIG1-deficient mice reveals increased short-term structural plasticity. Notably, the overall levels of PSD-95 or glutamate receptors enriched in postsynaptic biochemical fractions remain unaltered; however, activity-dependent synapse development is strongly compromised upon the loss of SynDIG1, supporting its importance for excitatory synapse maturation. Together, these data are consistent with a model in which SynDIG1 regulates the maturation of excitatory synapse structure and function in the mouse hippocampus in vivo.
Keywords: AMPA receptor; SynDIG1; excitatory synapse; hippocampus; synapse development; synapse maturation.
Figures
Similar articles
-
Disruption of Coordinated Presynaptic and Postsynaptic Maturation Underlies the Defects in Hippocampal Synapse Stability and Plasticity in Abl2/Arg-Deficient Mice.J Neurosci. 2016 Jun 22;36(25):6778-91. doi: 10.1523/JNEUROSCI.4092-15.2016. J Neurosci. 2016. PMID: 27335408 Free PMC article.
-
SynDIG1 regulation of excitatory synapse maturation.J Physiol. 2012 Jan 1;590(1):33-8. doi: 10.1113/jphysiol.2011.213884. Epub 2011 Aug 30. J Physiol. 2012. PMID: 21878521 Free PMC article. Review.
-
Activity-Dependent Palmitoylation Controls SynDIG1 Stability, Localization, and Function.J Neurosci. 2016 Jul 20;36(29):7562-8. doi: 10.1523/JNEUROSCI.4859-14.2016. J Neurosci. 2016. PMID: 27445135 Free PMC article.
-
SynDIG1: an activity-regulated, AMPA- receptor-interacting transmembrane protein that regulates excitatory synapse development.Neuron. 2010 Jan 14;65(1):80-93. doi: 10.1016/j.neuron.2009.12.021. Neuron. 2010. PMID: 20152115 Free PMC article.
-
Making of a Synapse: Recurrent Roles of Drebrin A at Excitatory Synapses Throughout Life.Adv Exp Med Biol. 2017;1006:119-139. doi: 10.1007/978-4-431-56550-5_8. Adv Exp Med Biol. 2017. PMID: 28865018 Review.
Cited by
-
Acute deletion of the central MR/GR steroid receptor correlates with changes in LTP, auditory neural gain, and GC-A cGMP signaling.Front Mol Neurosci. 2023 Feb 17;16:1017761. doi: 10.3389/fnmol.2023.1017761. eCollection 2023. Front Mol Neurosci. 2023. PMID: 36873102 Free PMC article.
-
The Na+-activated K+ channel Slack contributes to synaptic development and plasticity.Cell Mol Life Sci. 2021 Dec;78(23):7569-7587. doi: 10.1007/s00018-021-03953-0. Epub 2021 Oct 18. Cell Mol Life Sci. 2021. PMID: 34664085 Free PMC article.
-
Visualizing BDNF Transcript Usage During Sound-Induced Memory Linked Plasticity.Front Mol Neurosci. 2018 Jul 31;11:260. doi: 10.3389/fnmol.2018.00260. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30127717 Free PMC article.
-
Acyl-PEGyl Exchange Gel Shift Assay for Quantitative Determination of Palmitoylation of Brain Membrane Proteins.J Vis Exp. 2020 Mar 29;(157):10.3791/61018. doi: 10.3791/61018. J Vis Exp. 2020. PMID: 32281973 Free PMC article.
-
Interaction and Subcellular Association of PRRT1/SynDIG4 With AMPA Receptors.Front Synaptic Neurosci. 2021 Aug 2;13:705664. doi: 10.3389/fnsyn.2021.705664. eCollection 2021. Front Synaptic Neurosci. 2021. PMID: 34408636 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous