

Computational workflow for the fine-grained analysis of metagenomic samples
- PMID: 27801291
- PMCID: PMC5088524
- DOI: 10.1186/s12864-016-3063-x
Computational workflow for the fine-grained analysis of metagenomic samples
Abstract
Background: The field of metagenomics, defined as the direct genetic analysis of uncultured samples of genomes contained within an environmental sample, is gaining increasing popularity. The aim of studies of metagenomics is to determine the species present in an environmental community and identify changes in the abundance of species under different conditions. Current metagenomic analysis software faces bottlenecks due to the high computational load required to analyze complex samples.
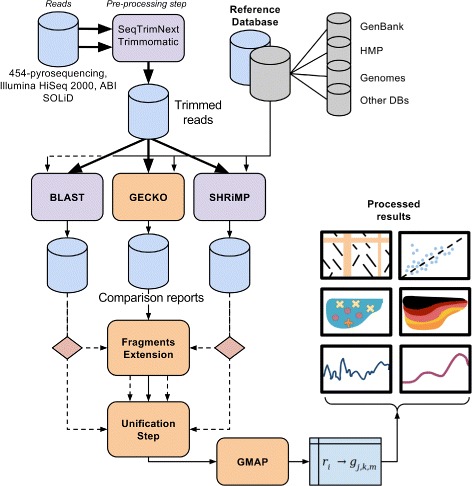
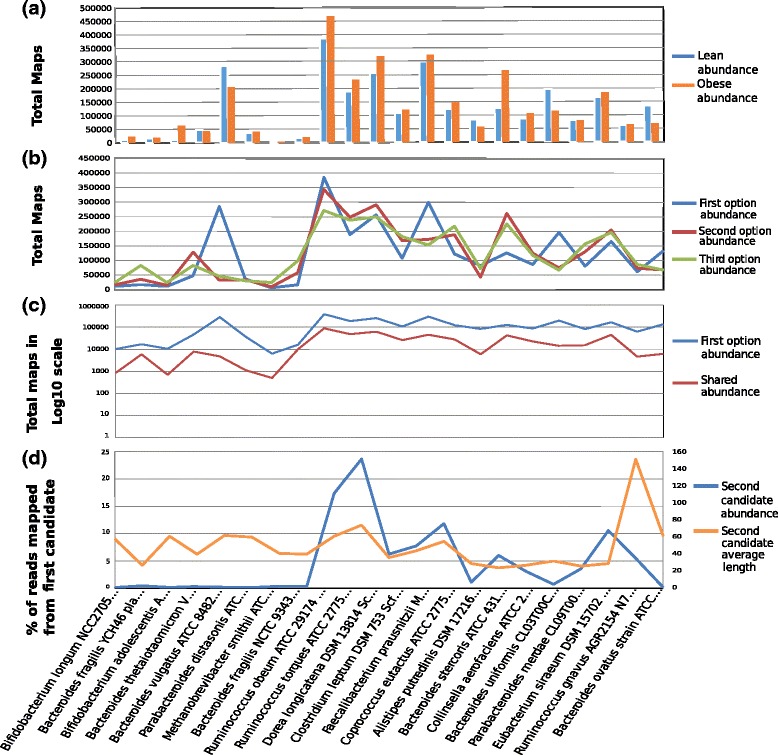
Results: A computational open-source workflow has been developed for the detailed analysis of metagenomes. This workflow provides new tools and datafile specifications that facilitate the identification of differences in abundance of reads assigned to taxa (mapping), enables the detection of reads of low-abundance bacteria (producing evidence of their presence), provides new concepts for filtering spurious matches, etc. Innovative visualization ideas for improved display of metagenomic diversity are also proposed to better understand how reads are mapped to taxa. Illustrative examples are provided based on the study of two collections of metagenomes from faecal microbial communities of adult female monozygotic and dizygotic twin pairs concordant for leanness or obesity and their mothers.
Conclusions: The proposed workflow provides an open environment that offers the opportunity to perform the mapping process using different reference databases. Additionally, this workflow shows the specifications of the mapping process and datafile formats to facilitate the development of new plugins for further post-processing. This open and extensible platform has been designed with the aim of enabling in-depth analysis of metagenomic samples and better understanding of the underlying biological processes.
Keywords: Annotational mapping; Differential abundance; Mapping over specific regions; Metagenome analysis; Open platform.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
