

An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography
- PMID: 27803195
- PMCID: PMC5088602
- DOI: 10.1101/gr.201863.115
An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography
Abstract
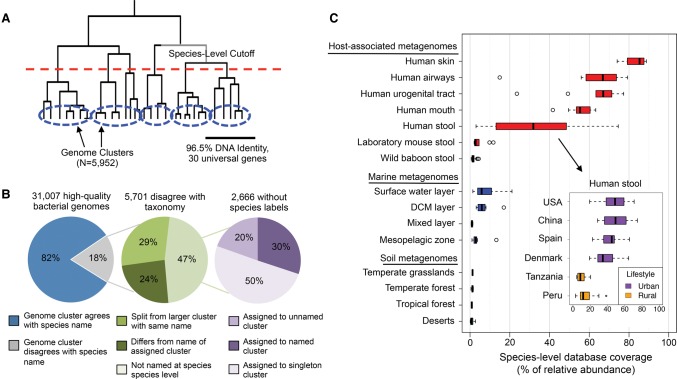
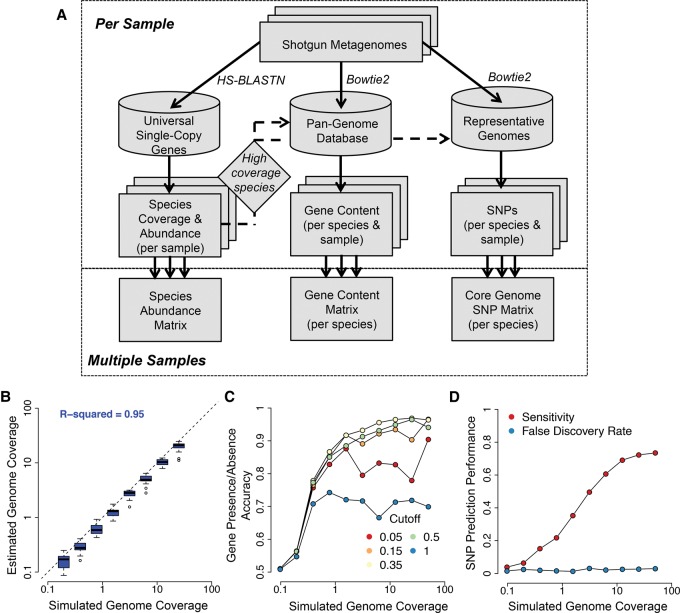
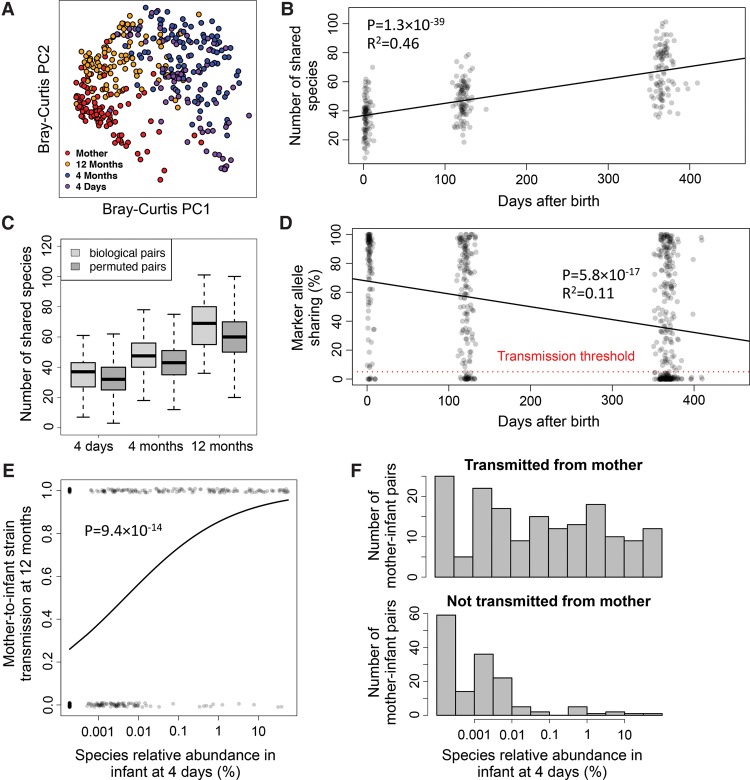
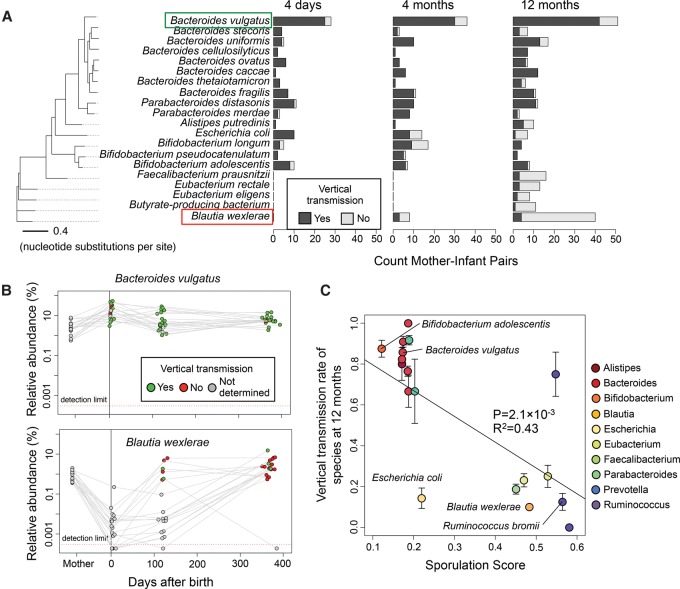
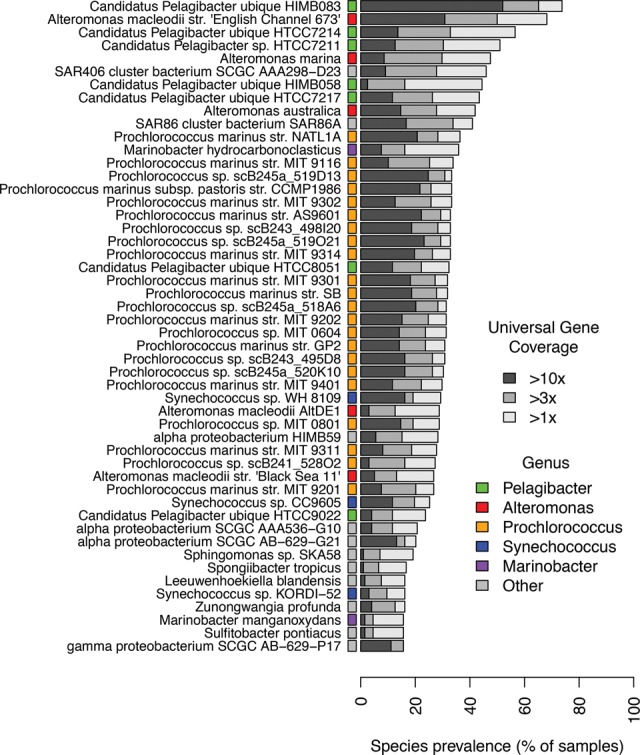
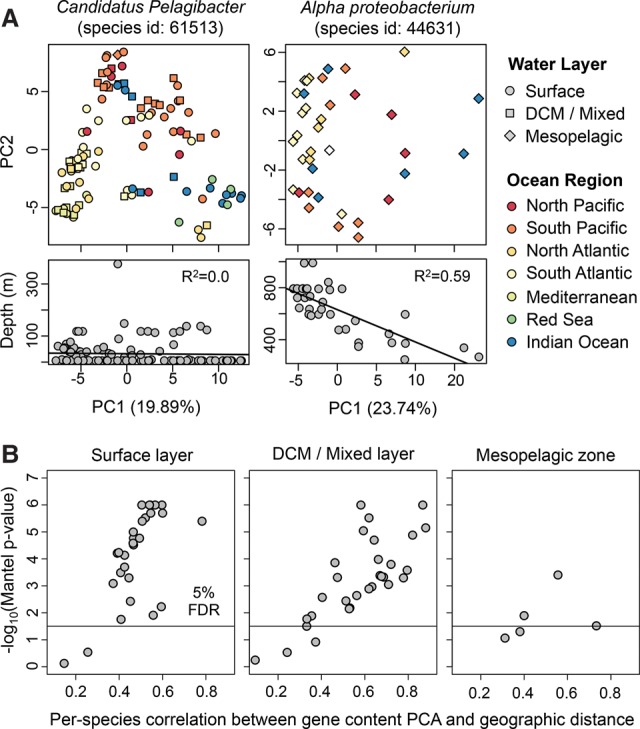
We present the Metagenomic Intra-species Diversity Analysis System (MIDAS), which is an integrated computational pipeline for quantifying bacterial species abundance and strain-level genomic variation, including gene content and single-nucleotide polymorphisms (SNPs), from shotgun metagenomes. Our method leverages a database of more than 30,000 bacterial reference genomes that we clustered into species groups. These cover the majority of abundant species in the human microbiome but only a small proportion of microbes in other environments, including soil and seawater. We applied MIDAS to stool metagenomes from 98 Swedish mothers and their infants over one year and used rare SNPs to track strains between hosts. Using this approach, we found that although species compositions of mothers and infants converged over time, strain-level similarity diverged. Specifically, early colonizing bacteria were often transmitted from an infant's mother, while late colonizing bacteria were often transmitted from other sources in the environment and were enriched for spore-formation genes. We also applied MIDAS to 198 globally distributed marine metagenomes and used gene content to show that many prevalent bacterial species have population structure that correlates with geographic location. Strain-level genetic variants present in metagenomes clearly reveal extensive structure and dynamics that are obscured when data are analyzed at a coarser taxonomic resolution.
© 2016 Nayfach et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. - PubMed
-
- Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. 2015. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17: 690–703. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources