

Molecular Pathophysiology of Congenital Long QT Syndrome
- PMID: 27807201
- PMCID: PMC5539372
- DOI: 10.1152/physrev.00008.2016
Molecular Pathophysiology of Congenital Long QT Syndrome
Abstract
Ion channels represent the molecular entities that give rise to the cardiac action potential, the fundamental cellular electrical event in the heart. The concerted function of these channels leads to normal cyclical excitation and resultant contraction of cardiac muscle. Research into cardiac ion channel regulation and mutations that underlie disease pathogenesis has greatly enhanced our knowledge of the causes and clinical management of cardiac arrhythmia. Here we review the molecular determinants, pathogenesis, and pharmacology of congenital Long QT Syndrome. We examine mechanisms of dysfunction associated with three critical cardiac currents that comprise the majority of congenital Long QT Syndrome cases: 1) IKs, the slow delayed rectifier current; 2) IKr, the rapid delayed rectifier current; and 3) INa, the voltage-dependent sodium current. Less common subtypes of congenital Long QT Syndrome affect other cardiac ionic currents that contribute to the dynamic nature of cardiac electrophysiology. Through the study of mutations that cause congenital Long QT Syndrome, the scientific community has advanced understanding of ion channel structure-function relationships, physiology, and pharmacological response to clinically employed and experimental pharmacological agents. Our understanding of congenital Long QT Syndrome continues to evolve rapidly and with great benefits: genotype-driven clinical management of the disease has improved patient care as precision medicine becomes even more a reality.
Copyright © 2017 the American Physiological Society.
Figures


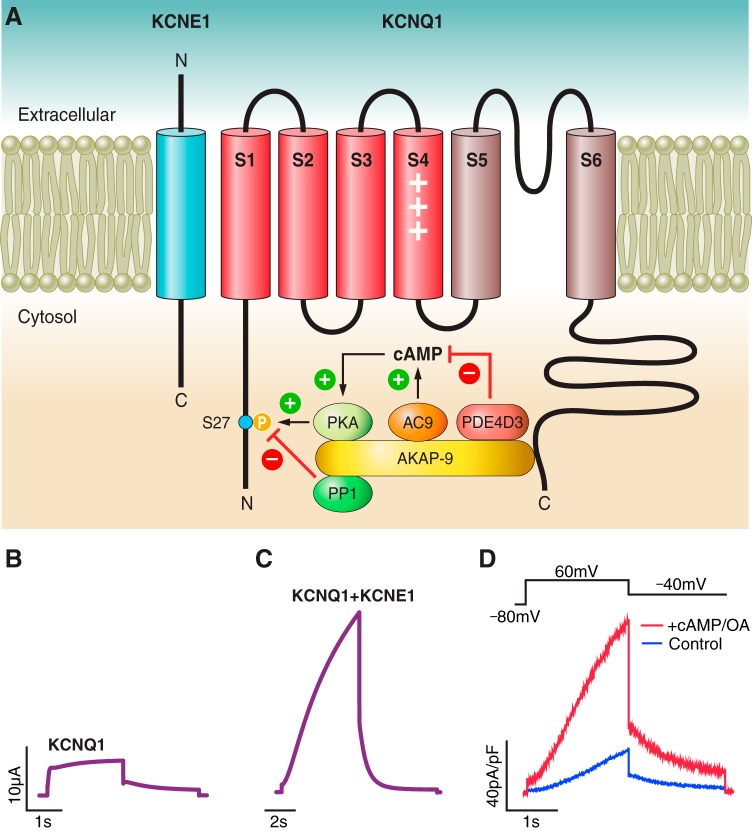
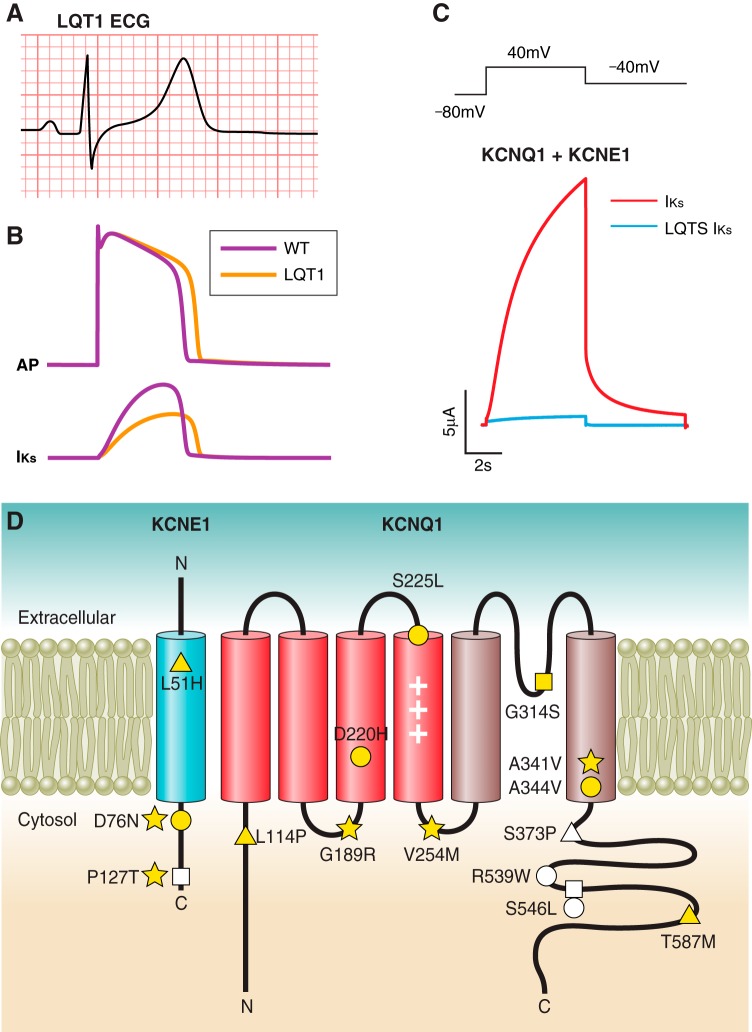


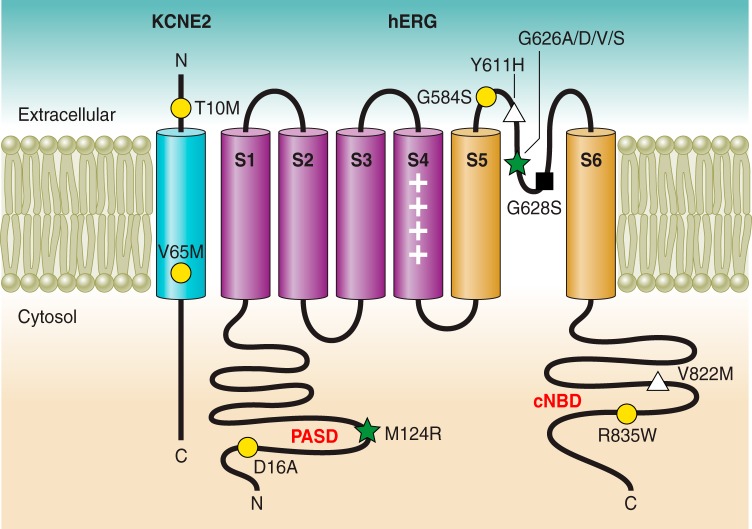
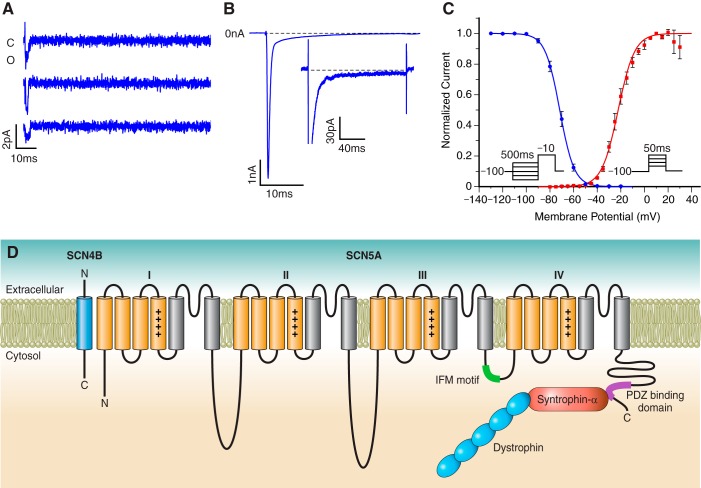
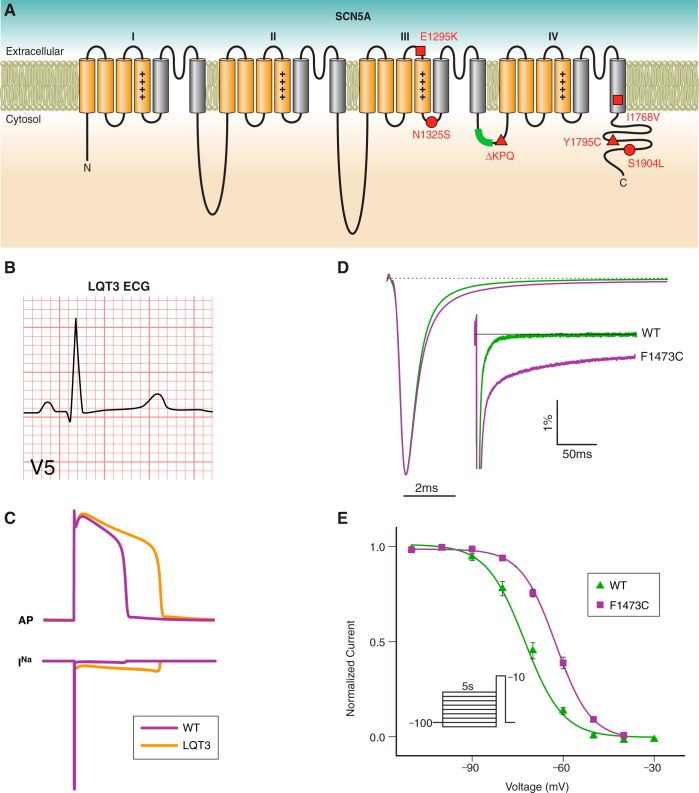

References
-
- Abbott GW. Biology of the KCNQ1 potassium channel. New J Sci 2014: 1–26, 2014.
-
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97: 175–187, 1999. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical