

Improving virus production through quasispecies genomic selection and molecular breeding
- PMID: 27808108
- PMCID: PMC5093897
- DOI: 10.1038/srep35962
Improving virus production through quasispecies genomic selection and molecular breeding
Abstract
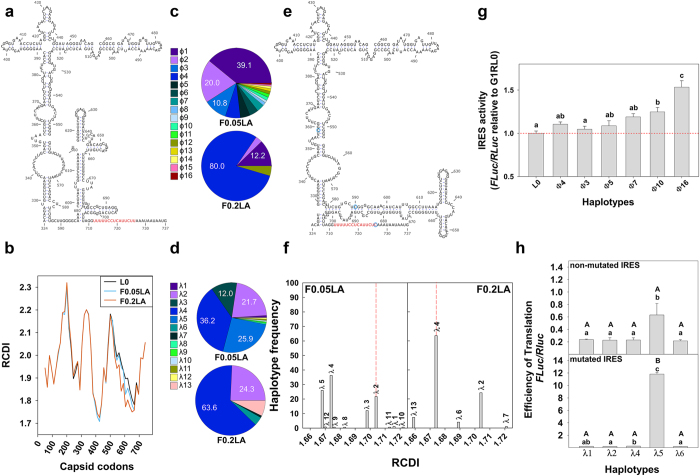
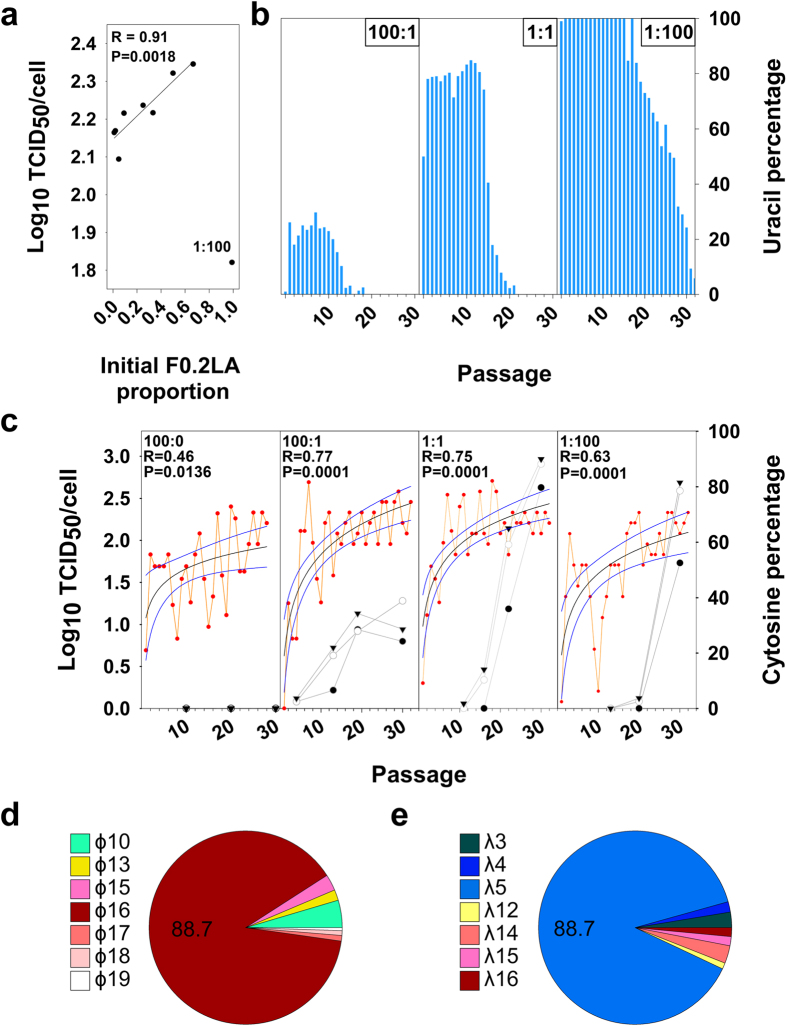
Virus production still is a challenging issue in antigen manufacture, particularly with slow-growing viruses. Deep-sequencing of genomic regions indicative of efficient replication may be used to identify high-fitness minority individuals suppressed by the ensemble of mutants in a virus quasispecies. Molecular breeding of quasispecies containing colonizer individuals, under regimes allowing more than one replicative cycle, is a strategy to select the fittest competitors among the colonizers. A slow-growing cell culture-adapted hepatitis A virus strain was employed as a model for this strategy. Using genomic selection in two regions predictive of efficient translation, the internal ribosome entry site and the VP1-coding region, high-fitness minority colonizer individuals were identified in a population adapted to conditions of artificially-induced cellular transcription shut-off. Molecular breeding of this population with a second one, also adapted to transcription shut-off and showing an overall colonizer phenotype, allowed the selection of a fast-growing population of great biotechnological potential.
Figures
References
-
- Lemon S. M., Whetter L., Chang K. H. & Brown E. A. Why do human hepatitis viruses replicate so poorly in cell cultures. FEMS Microbiol. Lett. 79, 455–459 (1992). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
