

A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish
- PMID: 27809318
- PMCID: PMC5630457
- DOI: 10.1038/nprot.2016.141
A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish
Abstract
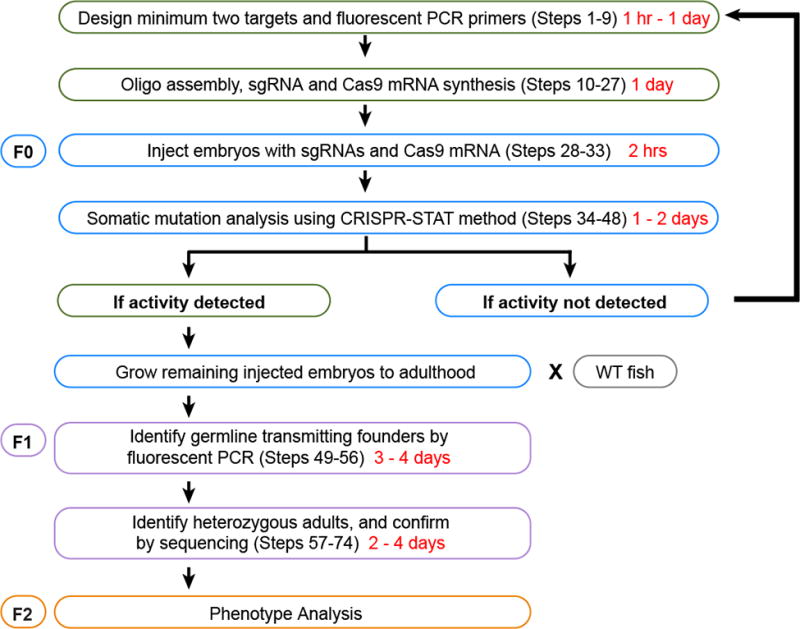
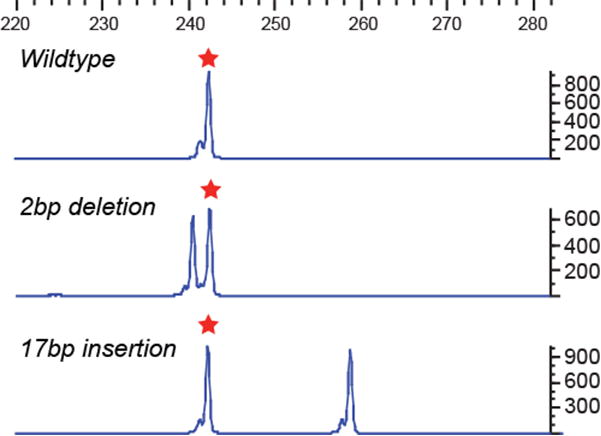
The zebrafish is a popular model organism for studying development and disease, and genetically modified zebrafish provide an essential tool for functional genomic studies. Numerous publications have demonstrated the efficacy of gene targeting in zebrafish using CRISPR/Cas9, and they have included descriptions of a variety of tools and methods for guide RNA synthesis and mutant identification. However, most of the published techniques are not readily scalable to increase throughput. We recently described a CRISPR/Cas9-based high-throughput mutagenesis and phenotyping pipeline in zebrafish. Here, we present a complete workflow for this pipeline, including target selection; cloning-free single-guide RNA (sgRNA) synthesis; microinjection; validation of the target-specific activity of the sgRNAs; founder screening to identify germline-transmitting mutations by fluorescence PCR; determination of the exact lesion by Sanger or next-generation sequencing (including software for analysis); and genotyping in the F1 or subsequent generations. Using these methods, sgRNAs can be evaluated in 3 d, zebrafish germline-transmitting mutations can be identified within 3 months and stable lines can be established within 6 months. Realistically, two researchers can target tens to hundreds of genes per year using this protocol.
Conflict of interest statement
Figures





Similar articles
-
High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9.Genome Res. 2015 Jul;25(7):1030-42. doi: 10.1101/gr.186379.114. Epub 2015 Jun 5. Genome Res. 2015. PMID: 26048245 Free PMC article.
-
Efficient identification of CRISPR/Cas9-induced insertions/deletions by direct germline screening in zebrafish.BMC Genomics. 2016 Mar 24;17:259. doi: 10.1186/s12864-016-2563-z. BMC Genomics. 2016. PMID: 27009152 Free PMC article.
-
CRISPR-Cas9-Mediated Genomic Deletions Protocol in Zebrafish.STAR Protoc. 2020 Dec 10;1(3):100208. doi: 10.1016/j.xpro.2020.100208. eCollection 2020 Dec 18. STAR Protoc. 2020. PMID: 33377102 Free PMC article.
-
New Developments in CRISPR/Cas-based Functional Genomics and their Implications for Research Using Zebrafish.Curr Gene Ther. 2017;17(4):286-300. doi: 10.2174/1566523217666171121164132. Curr Gene Ther. 2017. PMID: 29173171 Review.
-
Mutagenesis and phenotyping resources in zebrafish for studying development and human disease.Brief Funct Genomics. 2014 Mar;13(2):82-94. doi: 10.1093/bfgp/elt042. Epub 2013 Oct 26. Brief Funct Genomics. 2014. PMID: 24162064 Free PMC article. Review.
Cited by
-
Context-dependent hyperactivity in syngap1a and syngap1b zebrafish models of SYNGAP1-related disorder.Front Mol Neurosci. 2024 Jul 10;17:1401746. doi: 10.3389/fnmol.2024.1401746. eCollection 2024. Front Mol Neurosci. 2024. PMID: 39050824 Free PMC article.
-
Zebrafish models of human-duplicated SRGAP2 reveal novel functions in microglia and visual system development.bioRxiv [Preprint]. 2024 Sep 27:2024.09.11.612570. doi: 10.1101/2024.09.11.612570. bioRxiv. 2024. PMID: 39314374 Free PMC article. Preprint.
-
Modular genetic control of social status in a cichlid fish.Proc Natl Acad Sci U S A. 2020 Nov 10;117(45):28167-28174. doi: 10.1073/pnas.2008925117. Epub 2020 Oct 26. Proc Natl Acad Sci U S A. 2020. PMID: 33106426 Free PMC article.
-
Association of clinical severity with FANCB variant type in Fanconi anemia.Blood. 2020 Apr 30;135(18):1588-1602. doi: 10.1182/blood.2019003249. Blood. 2020. PMID: 32106311 Free PMC article.
-
High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system.Plant Biotechnol J. 2018 Jan;16(1):137-150. doi: 10.1111/pbi.12755. Epub 2017 Jun 20. Plant Biotechnol J. 2018. PMID: 28499063 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
