

Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes
- PMID: 27810900
- PMCID: PMC5207187
- DOI: 10.1074/jbc.M116.736991
Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes
Abstract
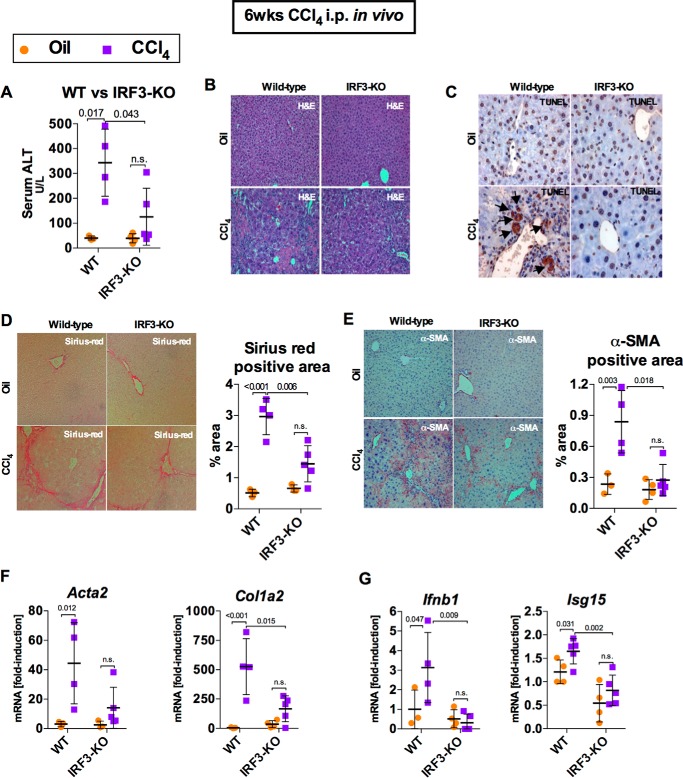
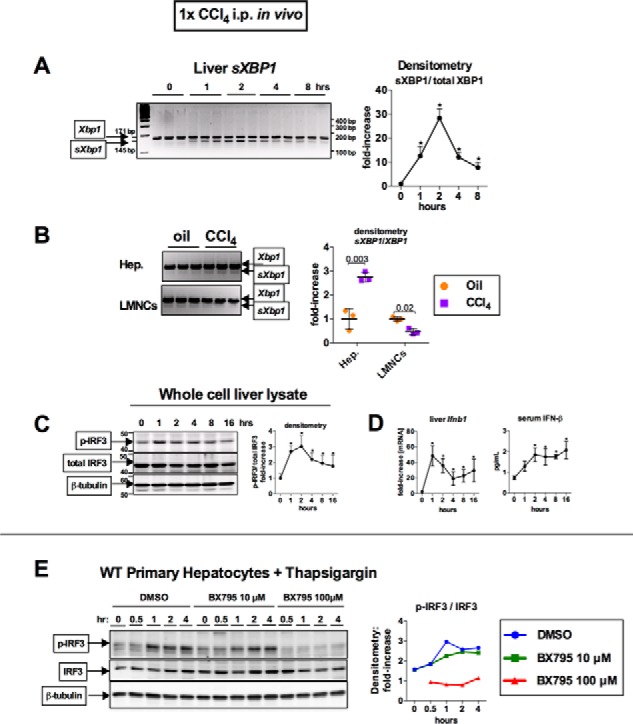
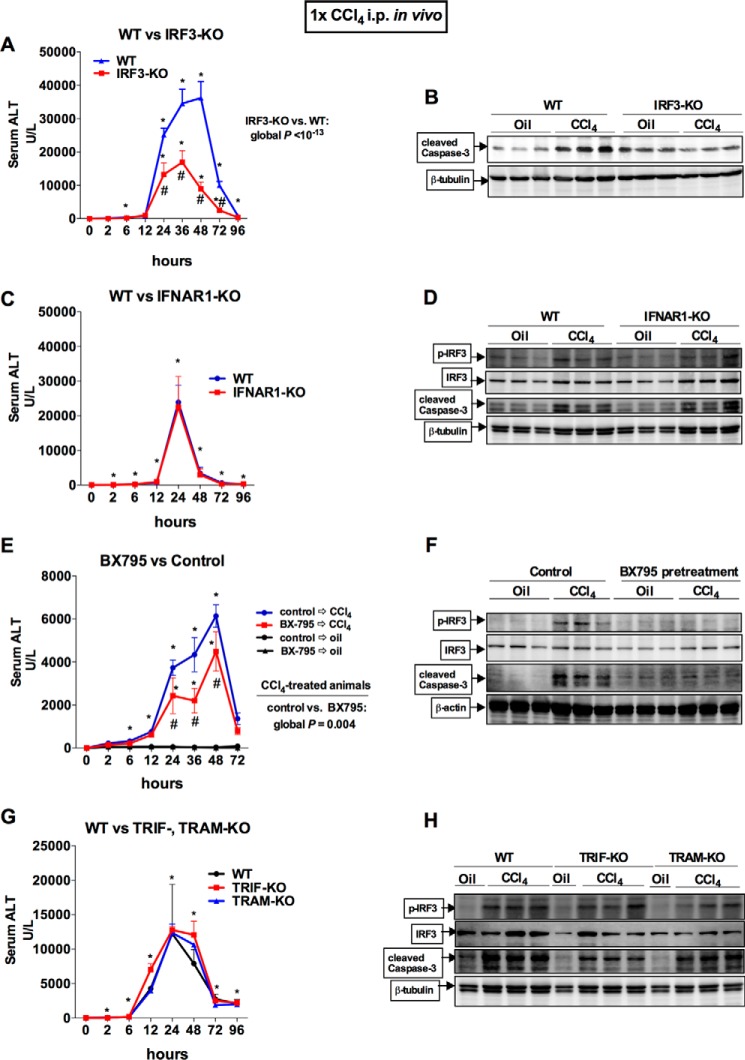
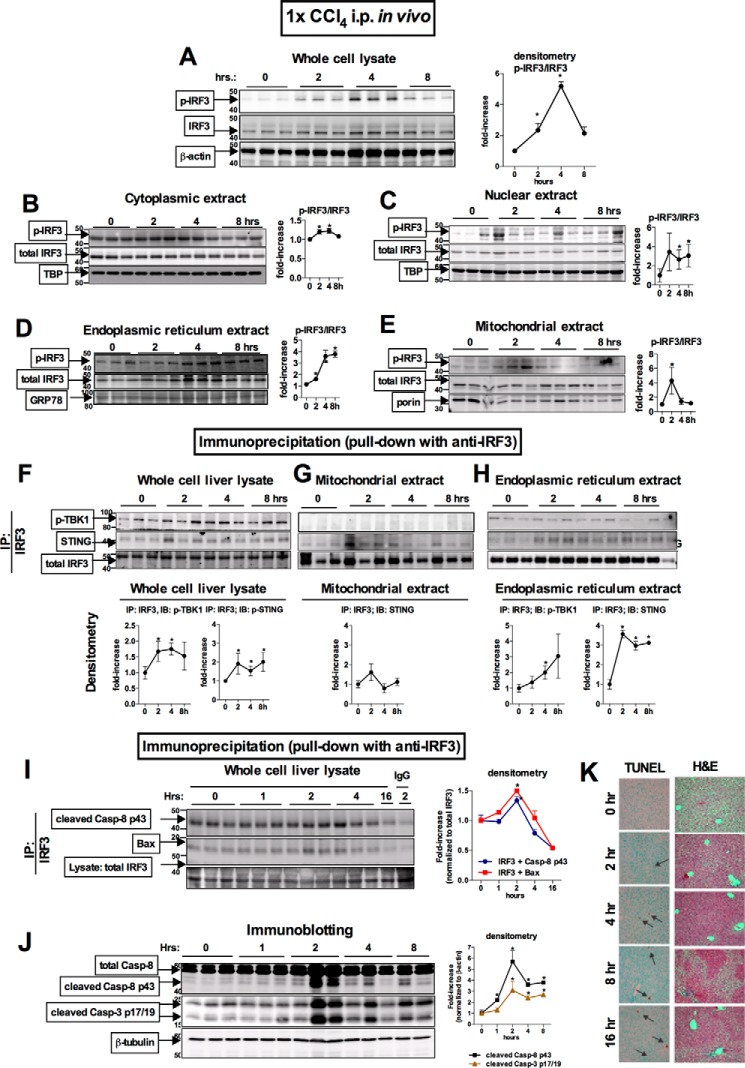
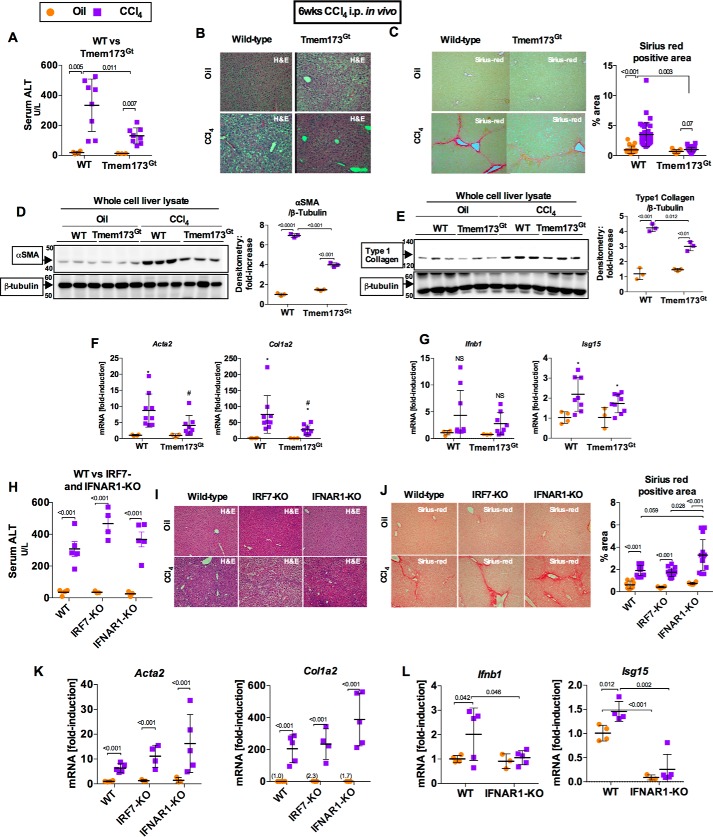
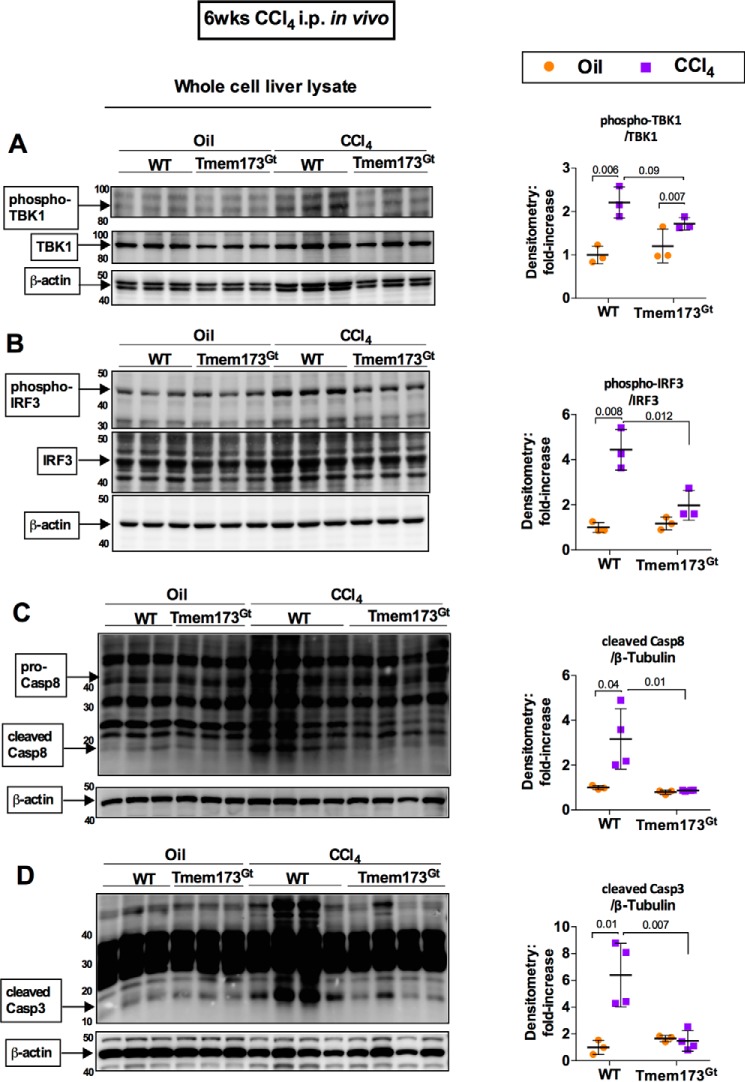
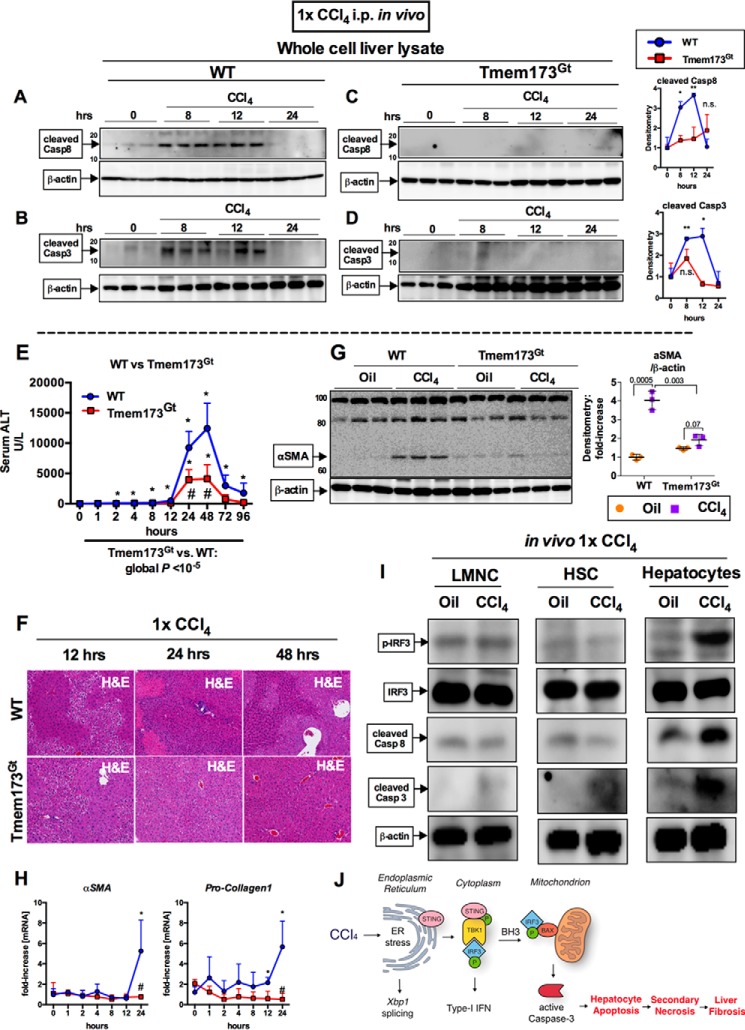
Fibrosis, driven by inflammation, marks the transition from benign to progressive stages of chronic liver diseases. Although inflammation promotes fibrogenesis, it is not known whether other events, such as hepatocyte death, are required for the development of fibrosis. Interferon regulatory factor 3 (IRF3) regulates hepatocyte apoptosis and production of type I IFNs. In the liver, IRF3 is activated via Toll-like receptor 4 (TLR4) signaling or the endoplasmic reticulum (ER) adapter, stimulator of interferon genes (STING). We hypothesized that IRF3-mediated hepatocyte death is an independent determinant of chemically induced liver fibrogenesis. To test this, we performed acute or chronic CCl4 administration to WT and IRF3-, Toll/Interleukin-1R (TIR) domain-containing adapter-inducing interferon-β (TRIF)-, TRIF-related adaptor molecule (TRAM)-, and STING-deficient mice. We report that acute CCl4 administration to WT mice resulted in early ER stress, activation of IRF3, and type I IFNs, followed by hepatocyte apoptosis and liver injury, accompanied by liver fibrosis upon repeated administration of CCl4 Deficiency of IRF3 or STING prevented hepatocyte death and fibrosis both in acute or chronic CCl4 In contrast, mice deficient in type I IFN receptors or in TLR4 signaling adaptors, TRAM or TRIF, upstream of IRF3, were not protected from hepatocyte death and/or fibrosis, suggesting that the pro-apoptotic role of IRF3 is independent of TLR signaling in fibrosis. Hepatocyte death is required for liver fibrosis with causal involvement of STING and IRF3. Thus, our results identify that IRF3, by its association with STING in the presence of ER stress, couples hepatocyte apoptosis with liver fibrosis and indicate that innate immune signaling regulates outcomes of liver fibrosis via modulation of hepatocyte death in the liver.
Keywords: apoptosis; endoplasmic reticulum stress (ER stress); fibrosis; interferon regulatory factor (IRF); liver injury.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Seki E., De Minicis S., Osterreicher C. H., Kluwe J., Osawa Y., Brenner D. A., and Schwabe R. F. (2007) TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 13, 1324–1332 - PubMed
-
- Rivera C. A., Bradford B. U., Hunt K. J., Adachi Y., Schrum L. W., Koop D. R., Burchardt E. R., Rippe R. A., and Thurman R. G. (2001) Attenuation of CCl4-induced hepatic fibrosis by GdCl3 treatment or dietary glycine. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G200–G207 - PubMed
-
- Danial N. N., and Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 - PubMed
-
- Jiang X., and Wang X. (2004) Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
