

--LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation
- PMID: 27810922
- PMCID: PMC5110014
- DOI: 10.1084/jem.20160041
--LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation
Abstract
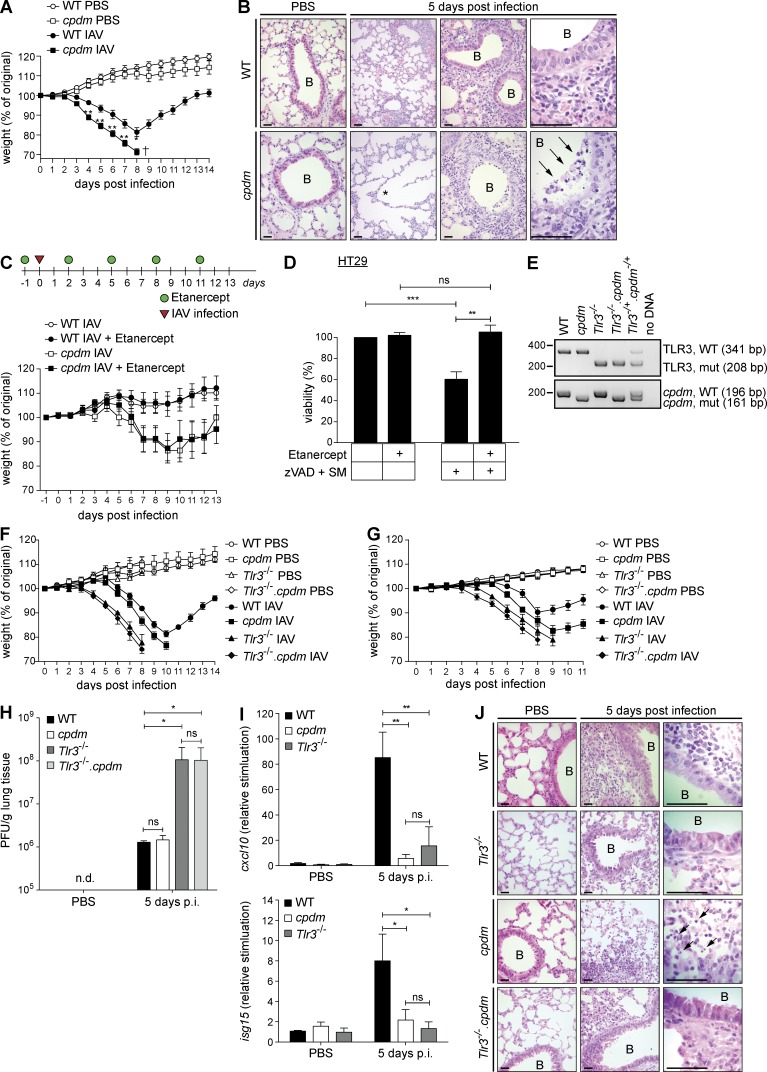
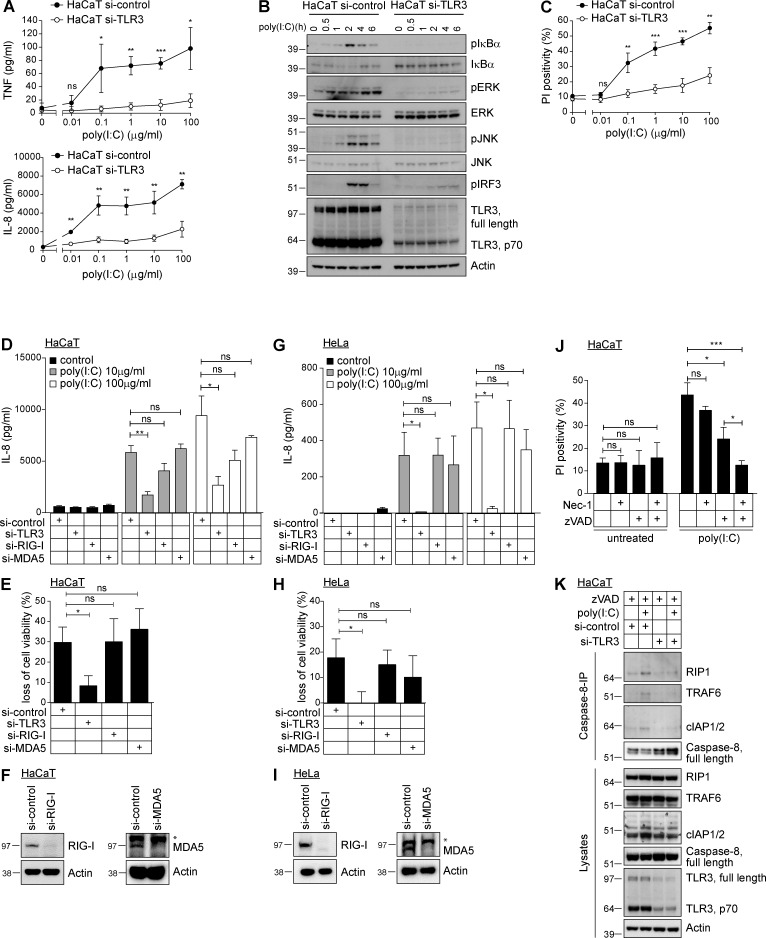
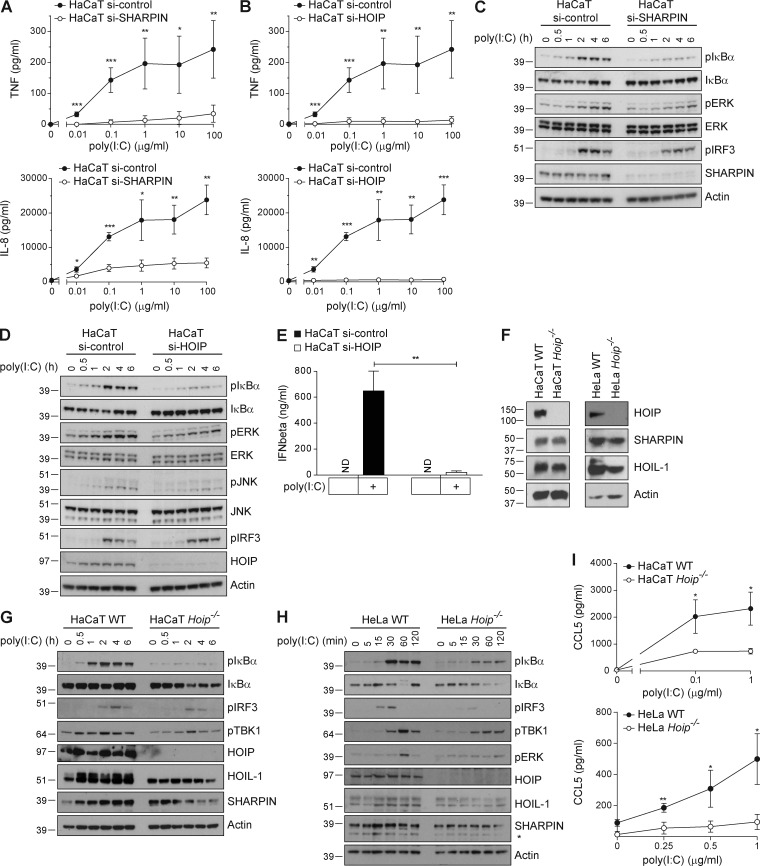
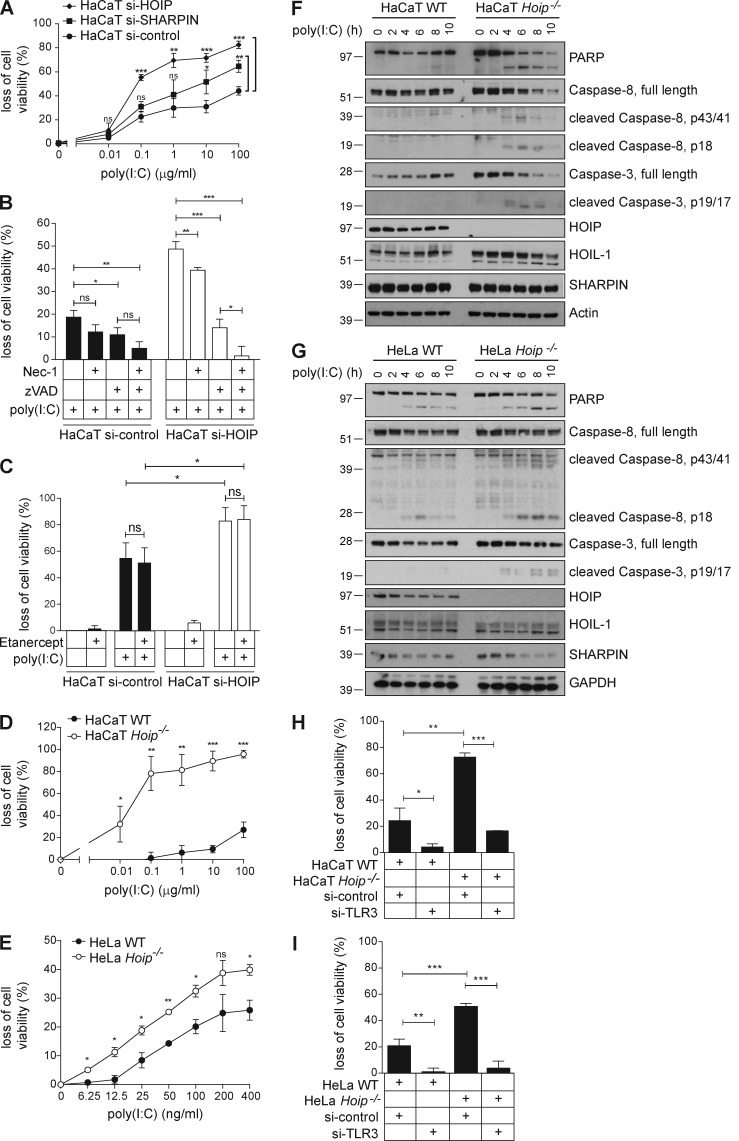
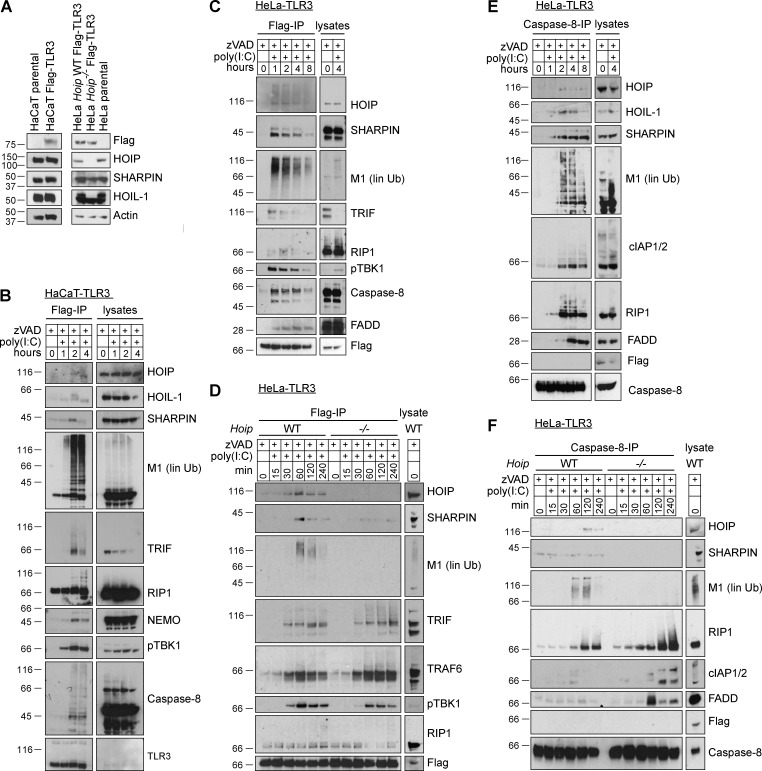
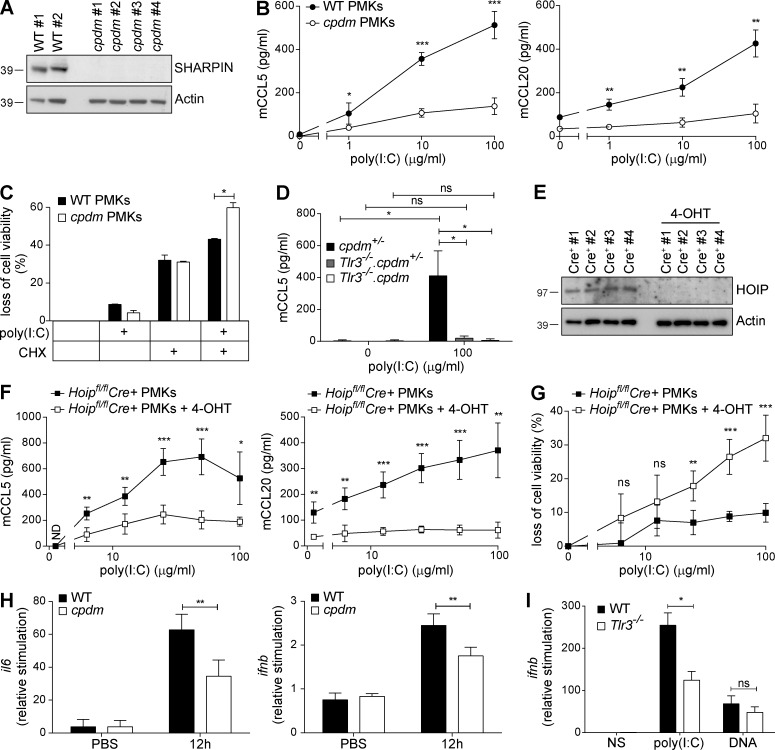
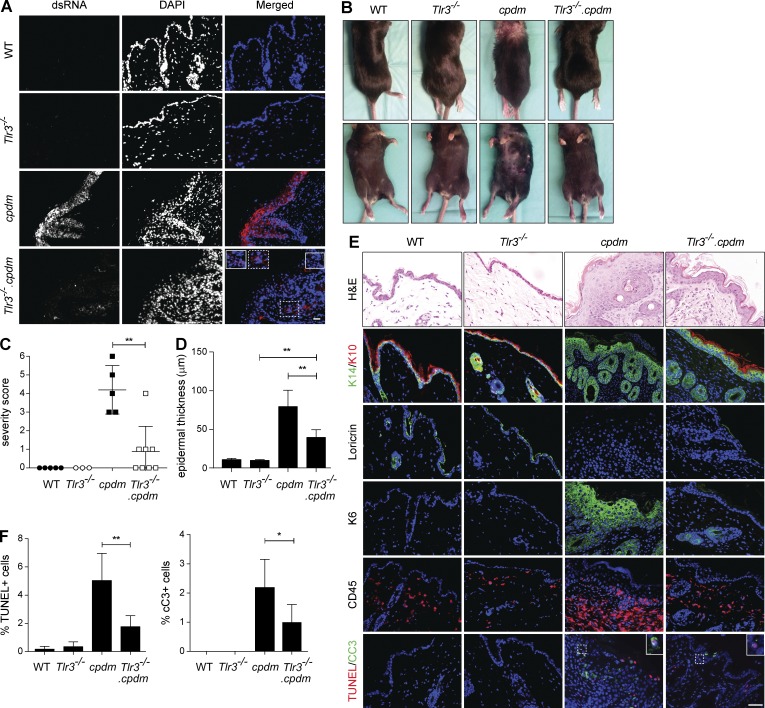
The linear ubiquitin chain assembly complex (LUBAC), consisting of SHANK-associated RH-domain-interacting protein (SHARPIN), heme-oxidized IRP2 ubiquitin ligase-1 (HOIL-1), and HOIL-1-interacting protein (HOIP), is a critical regulator of inflammation and immunity. This is highlighted by the fact that patients with perturbed linear ubiquitination caused by mutations in the Hoip or Hoil-1 genes, resulting in knockouts of these proteins, may simultaneously suffer from immunodeficiency and autoinflammation. TLR3 plays a crucial, albeit controversial, role in viral infection and tissue damage. We identify a pivotal role of LUBAC in TLR3 signaling and discover a functional interaction between LUBAC components and TLR3 as crucial for immunity to influenza A virus infection. On the biochemical level, we identify LUBAC components as interacting with the TLR3-signaling complex (SC), thereby enabling TLR3-mediated gene activation. Absence of LUBAC components increases formation of a previously unrecognized TLR3-induced death-inducing SC, leading to enhanced cell death. Intriguingly, excessive TLR3-mediated cell death, induced by double-stranded RNA present in the skin of SHARPIN-deficient chronic proliferative dermatitis mice (cpdm), is a major contributor to their autoinflammatory skin phenotype, as genetic coablation of Tlr3 substantially ameliorated cpdm dermatitis. Thus, LUBAC components control TLR3-mediated innate immunity, thereby preventing development of immunodeficiency and autoinflammation.
© 2016 Zinngrebe et al.
Figures
References
-
- Boisson B., Laplantine E., Prando C., Giliani S., Israelsson E., Xu Z., Abhyankar A., Israël L., Trevejo-Nunez G., Bogunovic D., et al. . 2012. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 13:1178–1186. 10.1038/ni.2457 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
