

ROS homeostasis and metabolism: a critical liaison for cancer therapy
- PMID: 27811934
- PMCID: PMC5133371
- DOI: 10.1038/emm.2016.119
ROS homeostasis and metabolism: a critical liaison for cancer therapy
Abstract
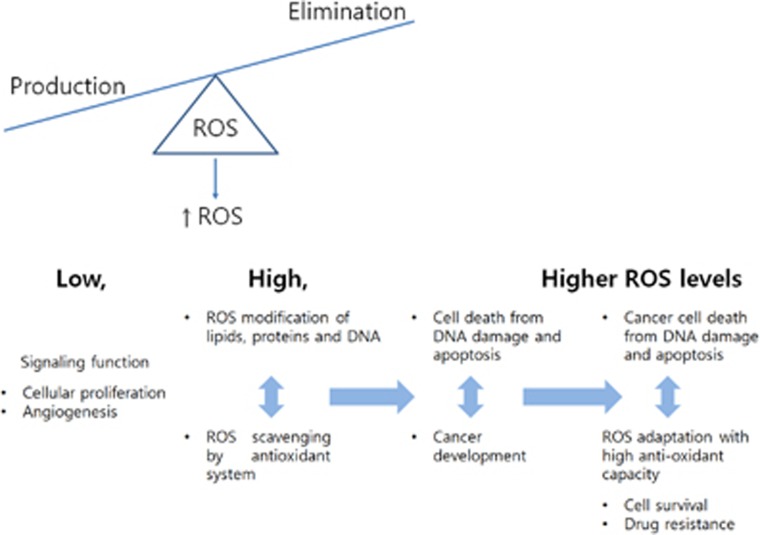
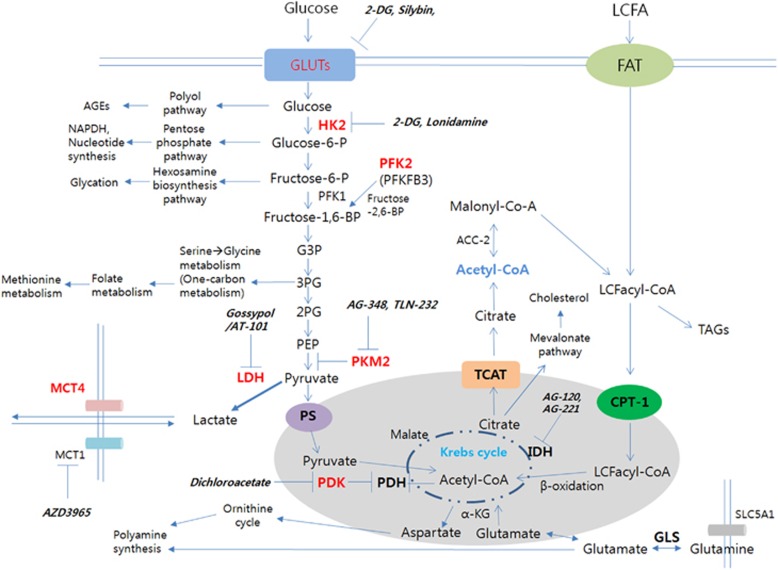
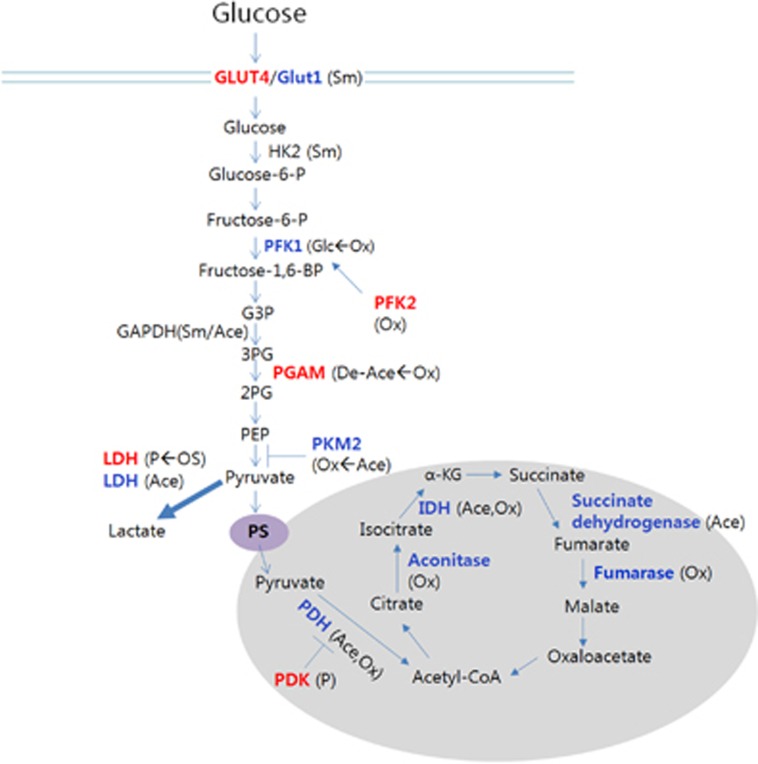
Evidence indicates that hypoxia and oxidative stress can control metabolic reprogramming of cancer cells and other cells in tumor microenvironments and that the reprogrammed metabolic pathways in cancer tissue can also alter the redox balance. Thus, important steps toward developing novel cancer therapy approaches would be to identify and modulate critical biochemical nodes that are deregulated in cancer metabolism and determine if the therapeutic efficiency can be influenced by changes in redox homeostasis in cancer tissues. In this review, we will explore the molecular mechanisms responsible for the metabolic reprogramming of tumor microenvironments, the functional modulation of which may disrupt the effects of or may be disrupted by redox homeostasis modulating cancer therapy.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
