

A Hidden Active Site in the Potential Drug Target Mycobacterium tuberculosis dUTPase Is Accessible through Small Amplitude Protein Conformational Changes
- PMID: 27815500
- PMCID: PMC5159494
- DOI: 10.1074/jbc.M116.734012
A Hidden Active Site in the Potential Drug Target Mycobacterium tuberculosis dUTPase Is Accessible through Small Amplitude Protein Conformational Changes
Abstract
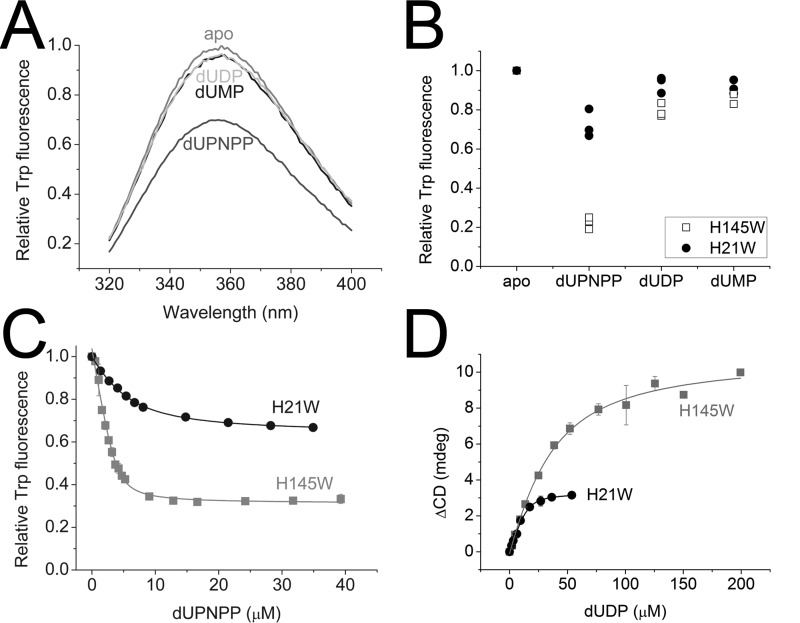
dUTPases catalyze the hydrolysis of dUTP into dUMP and pyrophosphate to maintain the proper nucleotide pool for DNA metabolism. Recent evidence suggests that dUTPases may also represent a selective drug target in mycobacteria because of the crucial role of these enzymes in maintaining DNA integrity. Nucleotide-hydrolyzing enzymes typically harbor a buried ligand-binding pocket at interdomain or intersubunit clefts, facilitating proper solvent shielding for the catalyzed reaction. The mechanism by which substrate binds this hidden pocket and product is released in dUTPases is unresolved because of conflicting crystallographic and spectroscopic data. We sought to resolve this conflict by using a combination of random acceleration molecular dynamics (RAMD) methodology and structural and biochemical methods to study the dUTPase from Mycobacterium tuberculosis In particular, the RAMD approach used in this study provided invaluable insights into the nucleotide dissociation process that reconciles all previous experimental observations. Specifically, our data suggest that nucleotide binding takes place as a small stretch of amino acids transiently slides away and partially uncovers the active site. The in silico data further revealed a new dUTPase conformation on the pathway to a relatively open active site. To probe this model, we developed the Trp21 reporter and collected crystallographic, spectroscopic, and kinetic data that confirmed the interaction of Trp21 with the active site shielding C-terminal arm, suggesting that the RAMD method is effective. In summary, our computational simulations and spectroscopic results support the idea that small loop movements in dUTPase allow the shuttlingof the nucleotides between the binding pocket and the solvent.
Keywords: Mycobacterium tuberculosis; X-ray crystallography; dUTPase; molecular dynamics; molecular modeling; nucleotide; pre-steady-state kinetics; solvent accessibility; substrate binding.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures











References
-
- Gai D., Zhao R., Li D., Finkielstein C. V., and Chen X. S. (2004) Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119, 47–60 - PubMed
-
- Velankar S. S., Soultanas P., Dillingham M. S., Subramanya H. S., and Wigley D. B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97, 75–84 - PubMed
-
- Sablin E. P., and Fletterick R. J. (2001) Nucleotide switches in molecular motors: structural analysis of kinesins and myosins. Curr. Opin Struct. Biol. 11, 716–724 - PubMed
-
- Shuman S., and Lima C. D. (2004) The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr. Opin Struct. Biol. 14, 757–764 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
