

Inferring Heterozygosity from Ancient and Low Coverage Genomes
- PMID: 27821432
- PMCID: PMC5223511
- DOI: 10.1534/genetics.116.189985
Inferring Heterozygosity from Ancient and Low Coverage Genomes
Abstract
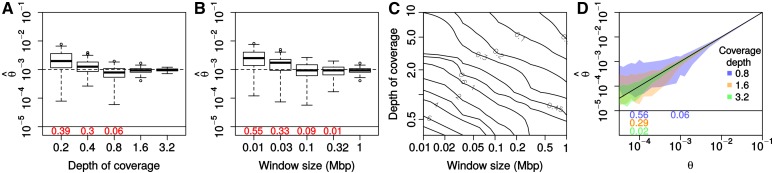
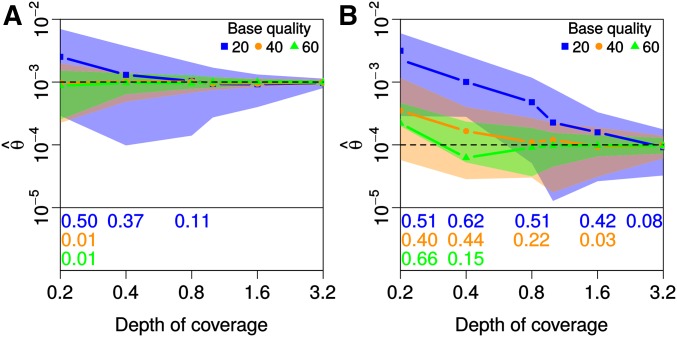
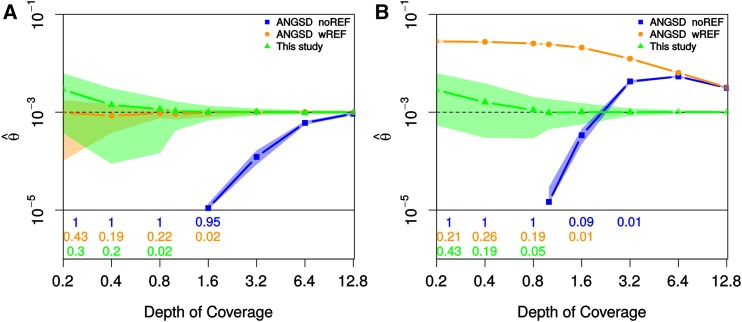
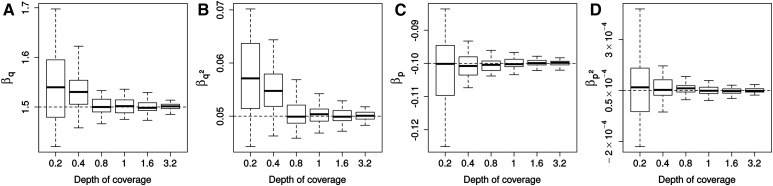
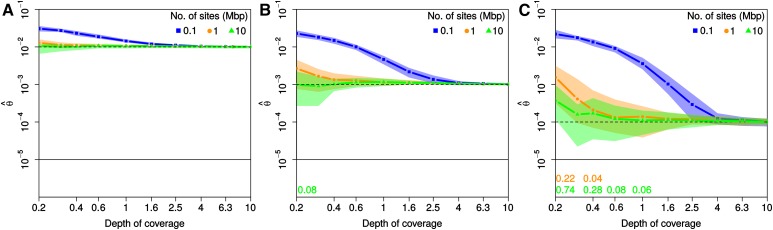
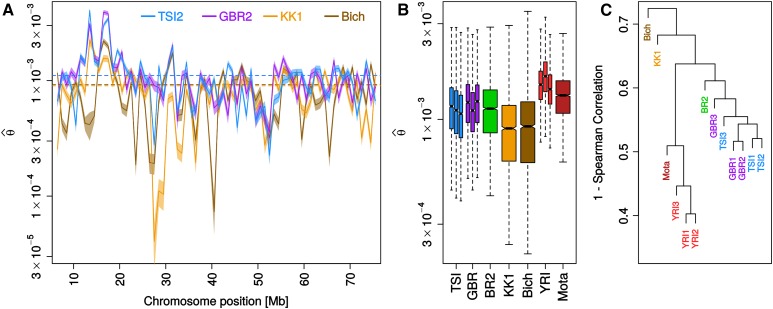
While genetic diversity can be quantified accurately from high coverage sequencing data, it is often desirable to obtain such estimates from data with low coverage, either to save costs or because of low DNA quality, as is observed for ancient samples. Here, we introduce a method to accurately infer heterozygosity probabilistically from sequences with average coverage [Formula: see text] of a single individual. The method relaxes the infinite sites assumption of previous methods, does not require a reference sequence, except for the initial alignment of the sequencing data, and takes into account both variable sequencing errors and potential postmortem damage. It is thus also applicable to nonmodel organisms and ancient genomes. Since error rates as reported by sequencing machines are generally distorted and require recalibration, we also introduce a method to accurately infer recalibration parameters in the presence of postmortem damage. This method does not require knowledge about the underlying genome sequence, but instead works with haploid data (e.g., from the X-chromosome from mammalian males) and integrates over the unknown genotypes. Using extensive simulations we show that a few megabasepairs of haploid data are sufficient for accurate recalibration, even at average coverages as low as [Formula: see text] At similar coverages, our method also produces very accurate estimates of heterozygosity down to [Formula: see text] within windows of about 1 Mbp. We further illustrate the usefulness of our approach by inferring genome-wide patterns of diversity for several ancient human samples, and we found that 3000-5000-year-old samples showed diversity patterns comparable to those of modern humans. In contrast, two European hunter-gatherer samples exhibited not only considerably lower levels of diversity than modern samples, but also highly distinct distributions of diversity along their genomes. Interestingly, these distributions were also very different between the two samples, supporting earlier conclusions of a highly diverse and structured population in Europe prior to the arrival of farming.
Keywords: ancient DNA; base recalibration; heterozygosity; low coverage; postmortem damage.
Copyright © 2017 by the Genetics Society of America.
Figures
References
-
- Dempster A. A., Laird N. N., Rubin D. D. B., 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B 39: 1–38.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
