

Disordered APP metabolism and neurovasculature in trauma and aging: Combined risks for chronic neurodegenerative disorders
- PMID: 27829172
- PMCID: PMC5315701
- DOI: 10.1016/j.arr.2016.11.003
Disordered APP metabolism and neurovasculature in trauma and aging: Combined risks for chronic neurodegenerative disorders
Abstract
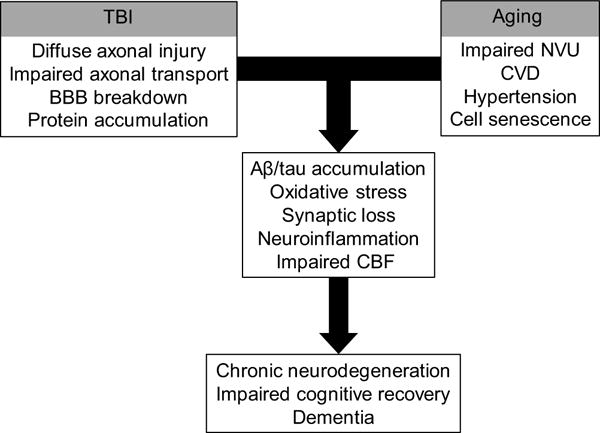
Traumatic brain injury (TBI), advanced age, and cerebral vascular disease are factors conferring increased risk for late onset Alzheimer's disease (AD). These conditions are also related pathologically through multiple interacting mechanisms. The hallmark pathology of AD consists of pathological aggregates of amyloid-β (Aβ) peptides and tau proteins. These molecules are also involved in neuropathology of several other chronic neurodegenerative diseases, and are under intense investigation in the aftermath of TBI as potential contributors to the risk for developing AD and chronic traumatic encephalopathy (CTE). The pathology of TBI is complex and dependent on injury severity, age-at-injury, and length of time between injury and neuropathological evaluation. In addition, the mechanisms influencing pathology and recovery after TBI likely involve genetic/epigenetic factors as well as additional disorders or comorbid states related to age and central and peripheral vascular health. In this regard, dysfunction of the aging neurovascular system could be an important link between TBI and chronic neurodegenerative diseases, either as a precipitating event or related to accumulation of AD-like pathology which is amplified in the context of aging. Thus with advanced age and vascular dysfunction, TBI can trigger self-propagating cycles of neuronal injury, pathological protein aggregation, and synaptic loss resulting in chronic neurodegenerative disease. In this review we discuss evidence supporting TBI and aging as dual, interacting risk factors for AD, and the role of Aβ and cerebral vascular dysfunction in this relationship. Evidence is discussed that Aβ is involved in cyto- and synapto-toxicity after severe TBI, and that its chronic effects are potentiated by aging and impaired cerebral vascular function. From a therapeutic perspective, we emphasize that in the fields of TBI- and aging-related neurodegeneration protective strategies should include preservation of neurovascular function.
Keywords: Aging; Alzheimer’s disease; Amyloid-beta; Brain trauma; Neurodegeneration; Neurovascular unit.
Published by Elsevier B.V.
Figures
Similar articles
-
Amyloid-beta and tau pathology following repetitive mild traumatic brain injury.Biochem Biophys Res Commun. 2017 Feb 19;483(4):1137-1142. doi: 10.1016/j.bbrc.2016.07.123. Epub 2016 Aug 1. Biochem Biophys Res Commun. 2017. PMID: 27492070 Review.
-
Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy?Exp Neurol. 2016 Jan;275 Pt 3(0 3):381-388. doi: 10.1016/j.expneurol.2015.06.015. Epub 2015 Jun 16. Exp Neurol. 2016. PMID: 26091850 Free PMC article. Review.
-
Brain injury-induced dysfunction of the blood brain barrier as a risk for dementia.Exp Neurol. 2020 Jun;328:113257. doi: 10.1016/j.expneurol.2020.113257. Epub 2020 Feb 21. Exp Neurol. 2020. PMID: 32092298 Review.
-
Traumatic Brain Injury Increases the Expression of Nos1, Aβ Clearance, and Epileptogenesis in APP/PS1 Mouse Model of Alzheimer's Disease.Mol Neurobiol. 2016 Dec;53(10):7010-7027. doi: 10.1007/s12035-015-9578-3. Epub 2015 Dec 15. Mol Neurobiol. 2016. PMID: 26671618
-
Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer's disease pathology.Prog Neurobiol. 2020 Feb;185:101730. doi: 10.1016/j.pneurobio.2019.101730. Epub 2019 Nov 25. Prog Neurobiol. 2020. PMID: 31778772
Cited by
-
Evaluation of Clinical Characteristics and CT Decision Rules in Elderly Patients with Minor Head Injury: A Prospective Multicenter Cohort Study.J Clin Med. 2023 Jan 27;12(3):982. doi: 10.3390/jcm12030982. J Clin Med. 2023. PMID: 36769631 Free PMC article.
-
Cutaneous impact location: a new tool to predict intracranial lesion among the elderly with mild traumatic brain injury?Scand J Trauma Resusc Emerg Med. 2020 Aug 31;28(1):87. doi: 10.1186/s13049-020-00781-2. Scand J Trauma Resusc Emerg Med. 2020. PMID: 32867809 Free PMC article.
-
Penetrating Ballistic-Like Brain Injury Leads to MicroRNA Dysregulation, BACE1 Upregulation, and Amyloid Precursor Protein Loss in Lesioned Rat Brain Tissues.Front Neurosci. 2020 Sep 18;14:915. doi: 10.3389/fnins.2020.00915. eCollection 2020. Front Neurosci. 2020. PMID: 33071724 Free PMC article.
-
Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases.Int J Mol Sci. 2024 Apr 18;25(8):4465. doi: 10.3390/ijms25084465. Int J Mol Sci. 2024. PMID: 38674050 Free PMC article. Review.
-
Subsequent Emergency Department Visits in Geriatric Mild Traumatic Brain Injury: Relationship with Fall, Payor, and Discharge Outcome.Healthcare (Basel). 2025 May 23;13(11):1236. doi: 10.3390/healthcare13111236. Healthcare (Basel). 2025. PMID: 40508849 Free PMC article.
References
-
- Abisambra JF, Scheff S. Brain injury in the context of tauopathies. J Alzheimers Dis. 2014;40:495–518. - PubMed
-
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood DG, Clark RSB, DeKosky ST. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp Neurol. 2006;197:437–450. - PubMed
-
- Abrahamson EE, Ikonomovic MD, Dixon CE, DeKosky ST. Simvastatin therapy prevents brain trauma-induced increases in beta-amyloid peptide levels. Ann Neurol. 2009;66:407–414. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
