

Copy number analysis reveals a novel multiexon deletion of the COLQ gene in congenital myasthenia
- PMID: 27830186
- PMCID: PMC5089432
- DOI: 10.1212/NXG.0000000000000117
Copy number analysis reveals a novel multiexon deletion of the COLQ gene in congenital myasthenia
Abstract
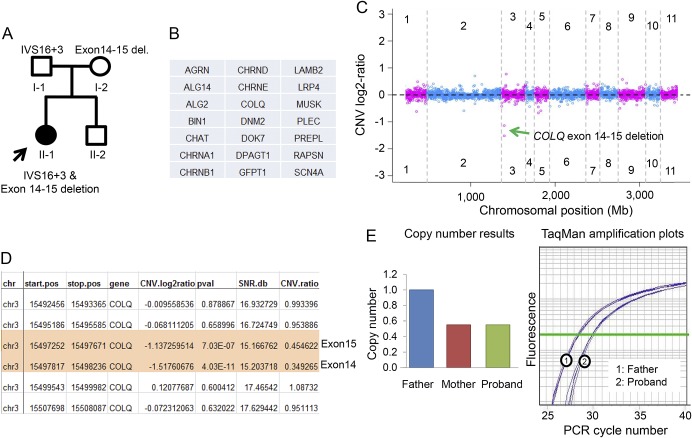
Congenital myasthenic syndrome (CMS) is genetically and clinically heterogeneous.1 Despite a considerable number of causal genes discovered, many patients are left without a specific diagnosis after genetic testing. The presumption is that novel genes yet to be discovered will account for the majority of such patients. However, it is also possible that we are neglecting a type of genetic variation: copy number changes (>50 bp) as causal for some of these patients. Next-generation sequencing (NGS) can simultaneously screen all known causal genes2 and is increasingly being validated to have a potential to identify copy number changes.3 We present a CMS case who did not receive a genetic diagnosis from previous Sanger sequencing, but through a novel copy number analysis algorithm integrated into our targeted NGS panel, we discovered a novel copy number mutation in the COLQ gene and made a genetic diagnosis. This discovery expands the genotype-phenotype correlation of CMS, leads to improved genetic counsel, and allows for specific pharmacologic treatment.1.
Figures
Similar articles
-
Two patients with congenital myasthenic syndrome caused by COLQ gene mutations and the consequent ColQ protein defect.Heliyon. 2023 Jan 26;9(2):e13272. doi: 10.1016/j.heliyon.2023.e13272. eCollection 2023 Feb. Heliyon. 2023. PMID: 36798769 Free PMC article.
-
Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes.Brain. 2008 Mar;131(Pt 3):747-59. doi: 10.1093/brain/awm325. Epub 2008 Jan 7. Brain. 2008. PMID: 18180250
-
Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation.BMJ Open. 2018 Oct 28;8(10):e021632. doi: 10.1136/bmjopen-2018-021632. BMJ Open. 2018. PMID: 30373780 Free PMC article.
-
[Congenital myasthenic syndromes: difficulties in the diagnosis, course and prognosis, and therapy--The French National Congenital Myasthenic Syndrome Network experience].Rev Neurol (Paris). 2013 Feb;169 Suppl 1:S45-55. doi: 10.1016/S0035-3787(13)70060-2. Rev Neurol (Paris). 2013. PMID: 23452772 Review. French.
-
[Congenital myasthenic syndromes: phenotypic expression and pathophysiological characterisation].Rev Neurol (Paris). 2004 Feb;160(2):163-76. doi: 10.1016/s0035-3787(04)70887-5. Rev Neurol (Paris). 2004. PMID: 15034473 Review. French.
Cited by
-
Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review.Int J Mol Sci. 2023 Feb 13;24(4):3730. doi: 10.3390/ijms24043730. Int J Mol Sci. 2023. PMID: 36835142 Free PMC article. Review.
-
Using gene panels in the diagnosis of neuromuscular disorders: A mini-review.Front Neurol. 2022 Oct 12;13:997551. doi: 10.3389/fneur.2022.997551. eCollection 2022. Front Neurol. 2022. PMID: 36313509 Free PMC article. Review.
-
The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein.Genes (Basel). 2020 Dec 18;11(12):1519. doi: 10.3390/genes11121519. Genes (Basel). 2020. PMID: 33353066 Free PMC article.
-
Novel copy number variation of COLQ gene in a Moroccan patient with congenital myasthenic syndrome: a case report and review of the literature.BMC Neurol. 2022 Aug 5;22(1):292. doi: 10.1186/s12883-022-02822-y. BMC Neurol. 2022. PMID: 35932018 Free PMC article. Review.
-
Intragenic DOK7 deletion detected by whole-genome sequencing in congenital myasthenic syndromes.Neurol Genet. 2017 May 3;3(3):e152. doi: 10.1212/NXG.0000000000000152. eCollection 2017 Jun. Neurol Genet. 2017. PMID: 28508085 Free PMC article.
References
-
- Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 2015;14:461. - PubMed
-
- Abicht A, Dusl M, Gallenmuller C, et al. . Congenital myasthenic syndromes: achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: a study of 680 patients. Hum Mutat 2012;33:1474–1484. - PubMed
-
- Ohno K, Brengman JM, Felice KJ, Cornblath DR, Engel AG. Congenital end-plate acetylcholinesterase deficiency caused by a nonsense mutation and an A-->G splice-donor-site mutation at position +3 of the collagenlike-tail-subunit gene (COLQ): how does G at position +3 result in aberrant splicing? Am J Hum Genet 1999;65:635–644. - PMC - PubMed
-
- Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet 2015;16:172–183. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources