

Decoding ALS: from genes to mechanism
- PMID: 27830784
- PMCID: PMC5585017
- DOI: 10.1038/nature20413
Decoding ALS: from genes to mechanism
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive and uniformly fatal neurodegenerative disease. A plethora of genetic factors have been identified that drive the degeneration of motor neurons in ALS, increase susceptibility to the disease or influence the rate of its progression. Emerging themes include dysfunction in RNA metabolism and protein homeostasis, with specific defects in nucleocytoplasmic trafficking, the induction of stress at the endoplasmic reticulum and impaired dynamics of ribonucleoprotein bodies such as RNA granules that assemble through liquid-liquid phase separation. Extraordinary progress in understanding the biology of ALS provides new reasons for optimism that meaningful therapies will be identified.
Figures
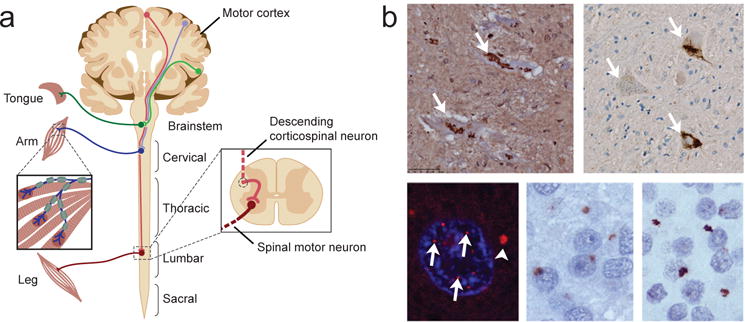
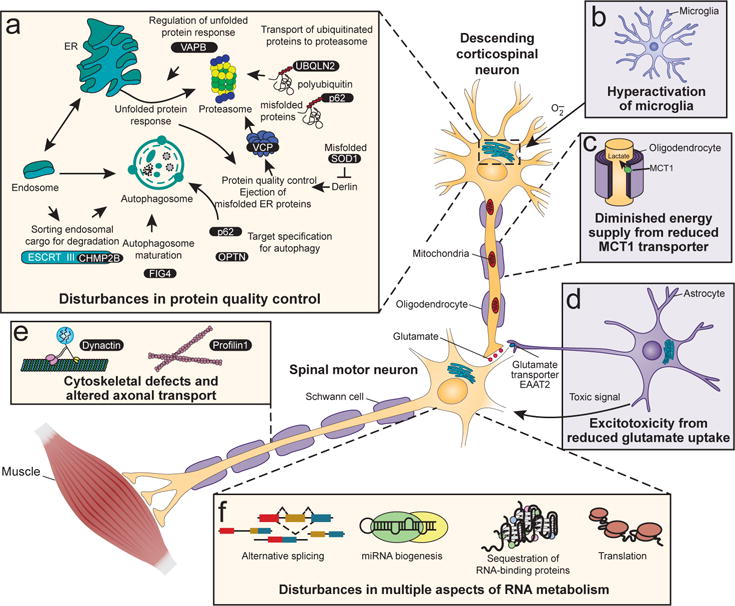
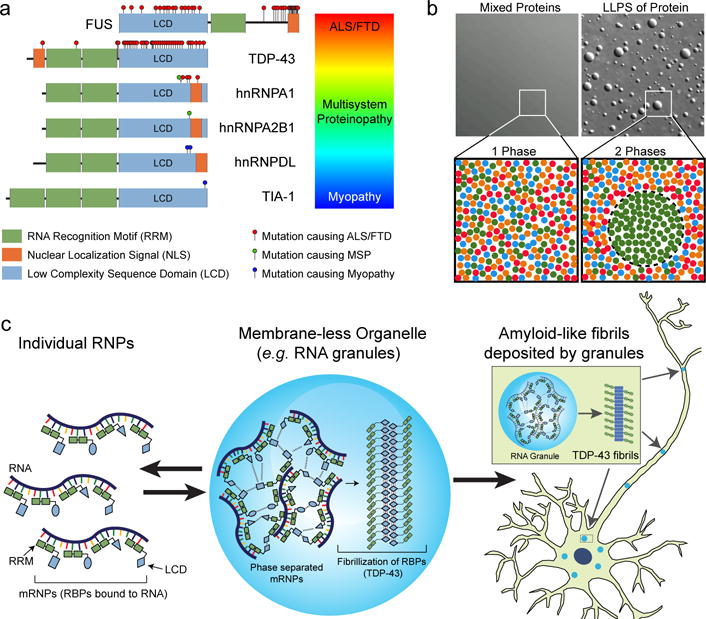
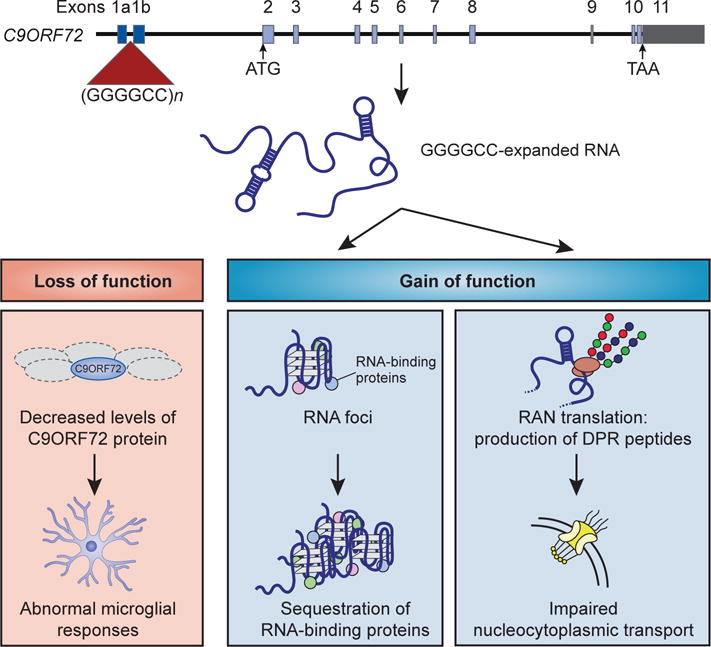
References
-
- Gamez J, et al. Mutational analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: should all sporadic ALS cases also be screened for SOD1? J Neurol Sci. 2006;247:21–28. - PubMed
-
- Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. This is the first report of a relationship between microsatellite expansion in ataxin-2 and ALS susceptibility. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
