

Tissue-type plasminogen activator controls neuronal death by raising surface dynamics of extrasynaptic NMDA receptors
- PMID: 27831563
- PMCID: PMC5260909
- DOI: 10.1038/cddis.2016.279
Tissue-type plasminogen activator controls neuronal death by raising surface dynamics of extrasynaptic NMDA receptors
Abstract
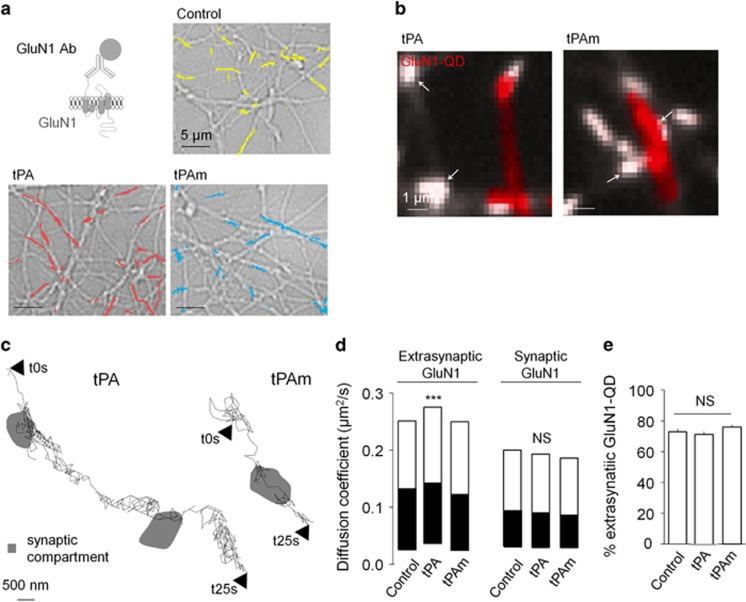
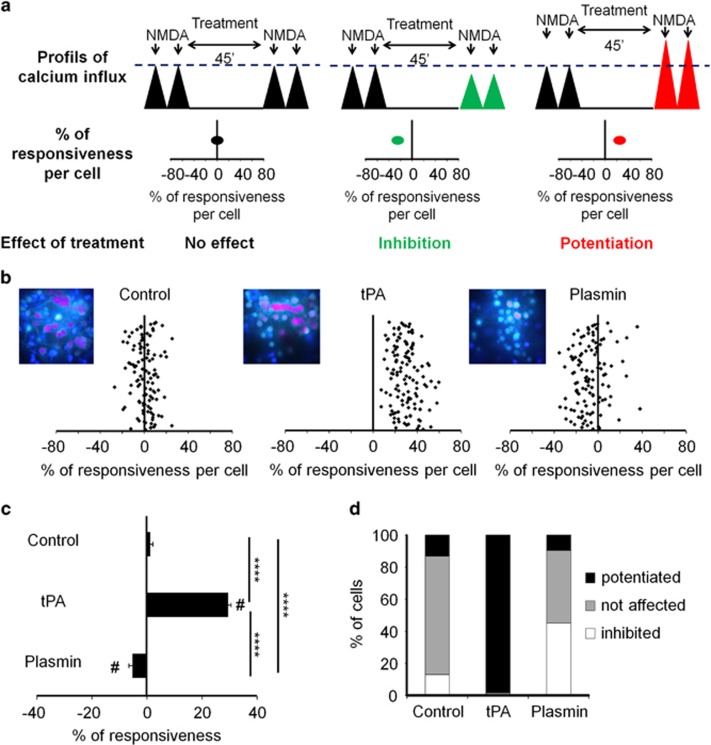
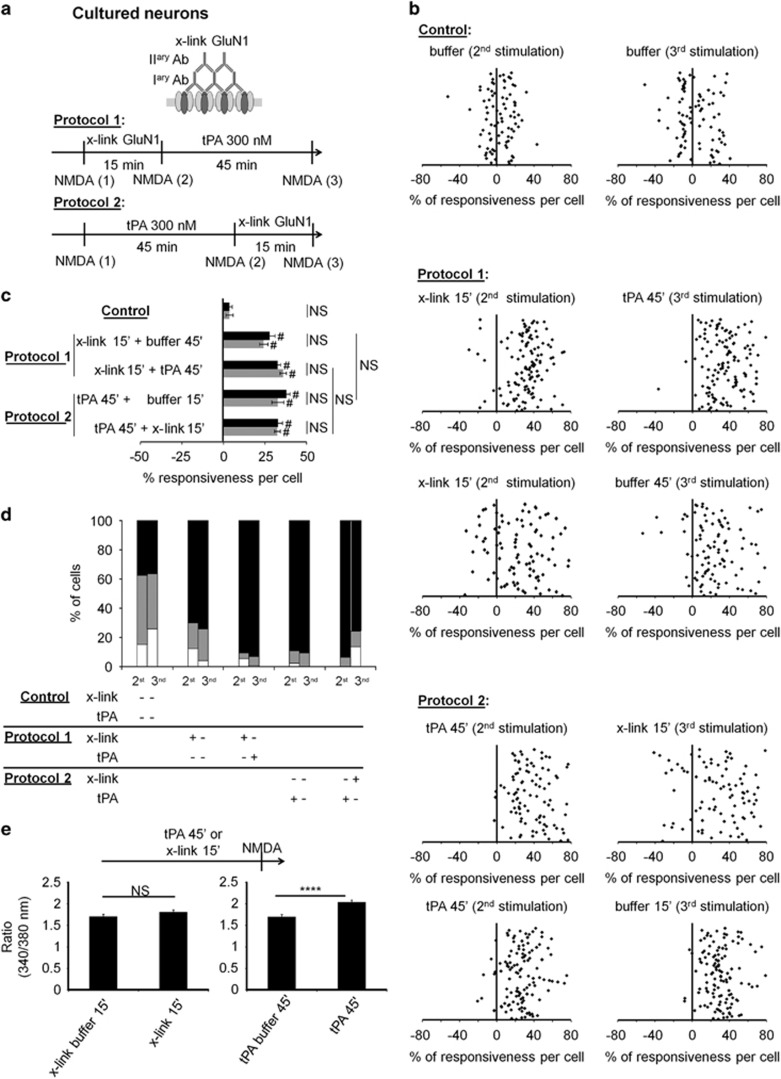
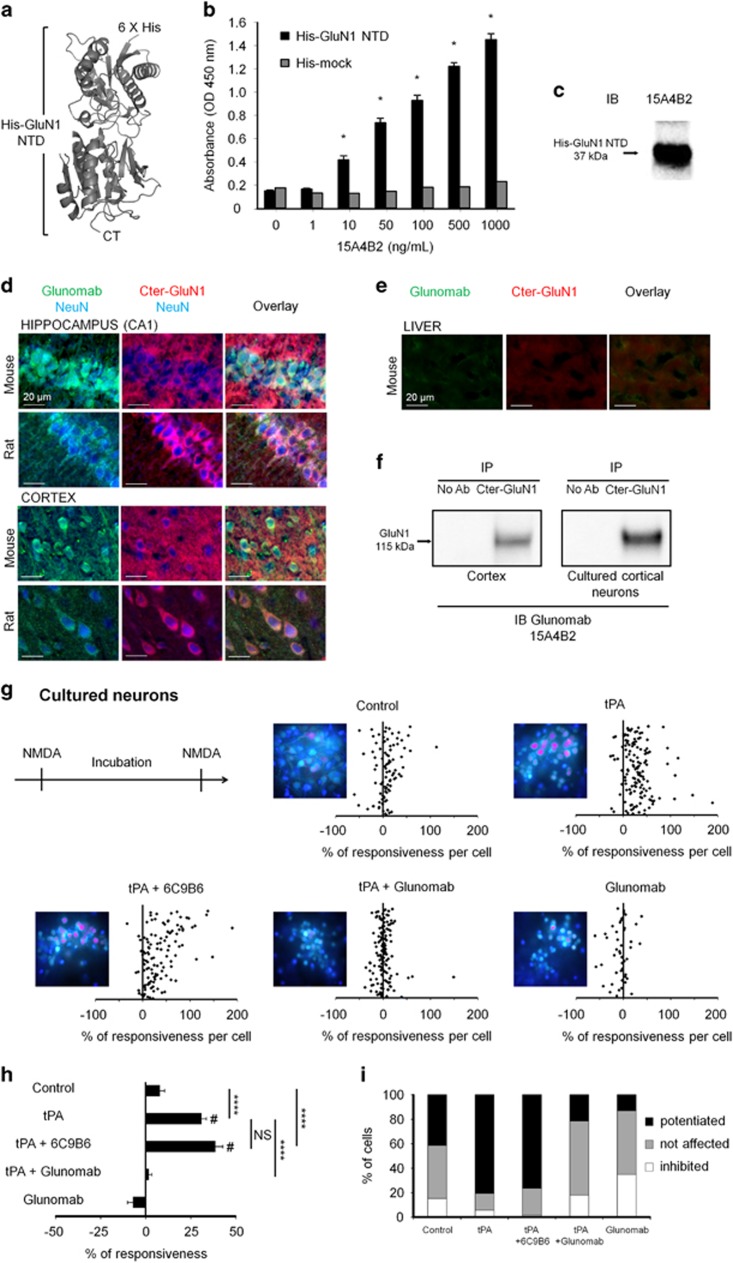
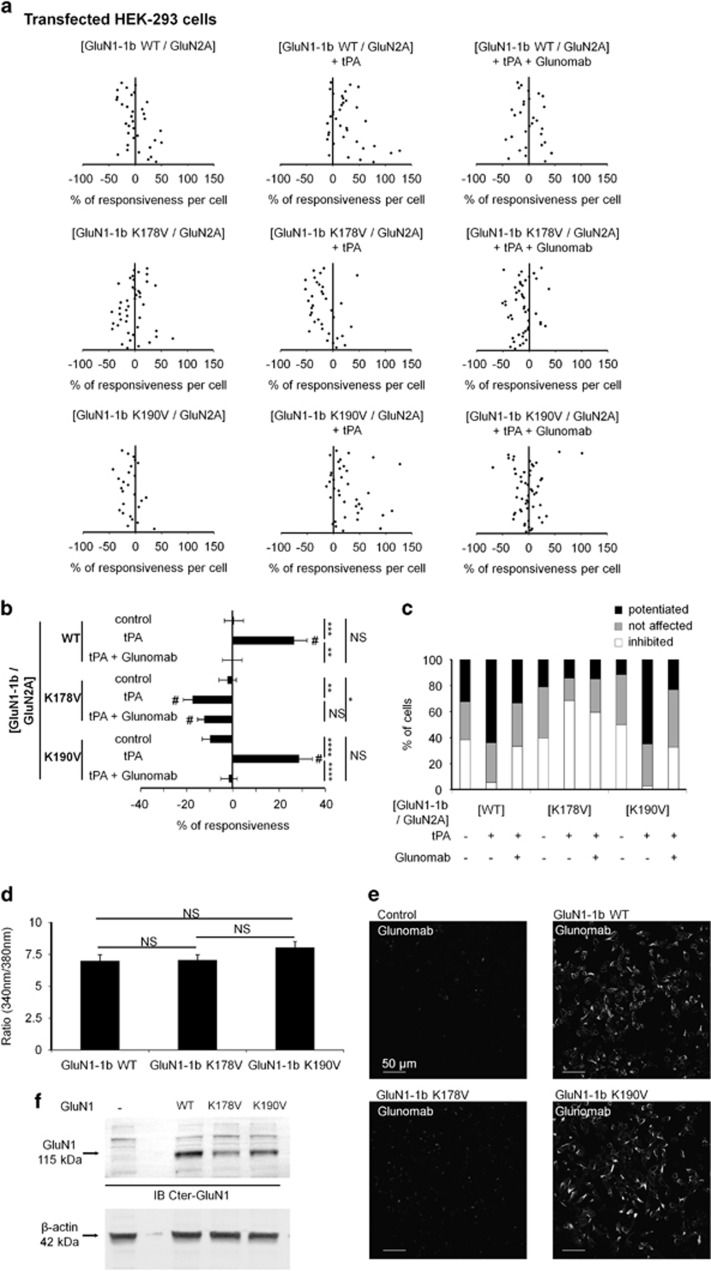
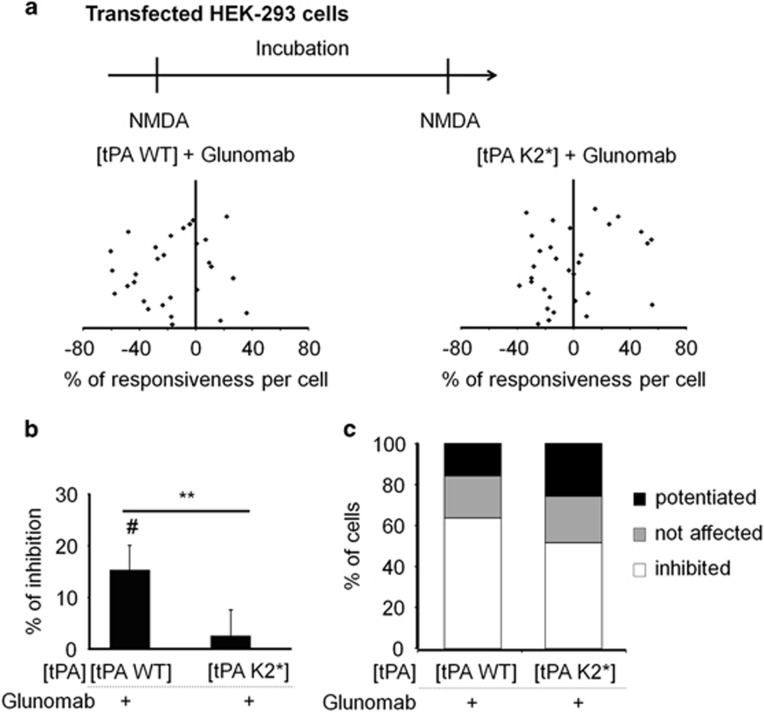
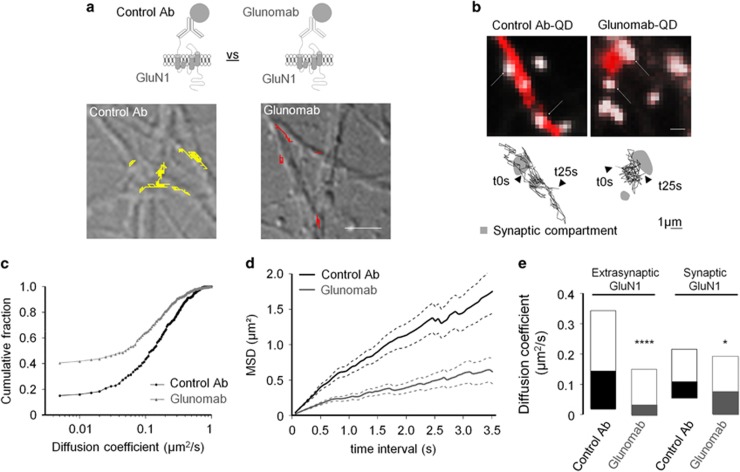
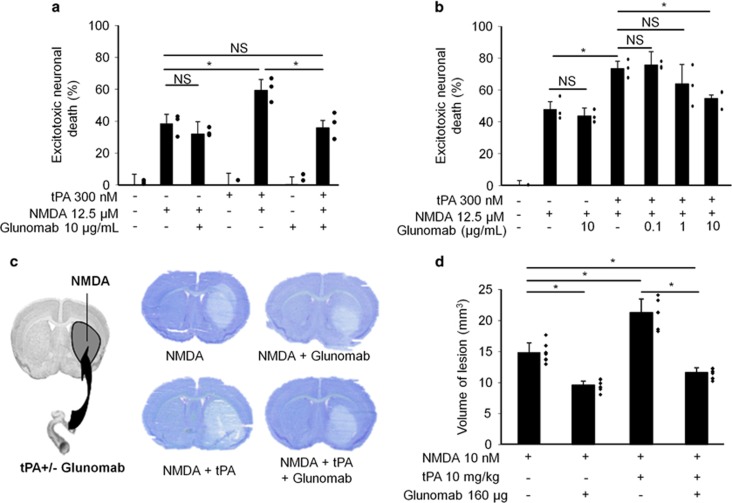
N-methyl-d-aspartate receptors (NMDARs) are ion channels whose synaptic versus extrasynaptic localization critically influences their functions. This distribution of NMDARs is highly dependent on their lateral diffusion at the cell membrane. Each obligatory subunit of NMDARs (GluN1 and GluN2) contains two extracellular clamshell-like domains with an agonist-binding domain and a distal N-terminal domain (NTD). To date, the roles and dynamics of the NTD of the GluN1 subunit in NMDAR allosteric signaling remain poorly understood. Using single nanoparticle tracking in mouse neurons, we demonstrate that the extracellular neuronal protease tissue-type plasminogen activator (tPA), well known to have a role in the synaptic plasticity and neuronal survival, leads to a selective increase of the surface dynamics and subsequent diffusion of extrasynaptic NMDARs. This process explains the previously reported ability of tPA to promote NMDAR-mediated calcium influx. In parallel, we developed a monoclonal antibody capable of specifically blocking the interaction of tPA with the NTD of the GluN1 subunit of NMDAR. Using this original approach, we demonstrate that the tPA binds the NTD of the GluN1 subunit at a lysine in position 178. Accordingly, when applied to mouse neurons, our selected antibody (named Glunomab) leads to a selective reduction of the tPA-mediated surface dynamics of extrasynaptic NMDARs, subsequent signaling and neurotoxicity, both in vitro and in vivo. Altogether, we demonstrate that the tPA is a ligand of the NTD of the obligatory GluN1 subunit of NMDAR acting as a modulator of their dynamic distribution at the neuronal surface and subsequent signaling.
Conflict of interest statement
DV, CA, K-UP and RM are co-inventors on the following patent 'Treatment of neurological or neurodegenerative disorders' (WO2011023250A1). The original patent W02011023250A1 was completed and now published under the number W02014187879A2 (additional references: EP2805972A1, WO2014187879A3). The present invention relates to the field of antibodies. In particular, it provides an anti-NMDA antibody or fragment or derivative thereof which is effective in inhibiting the deleterious effects of tissue-type plasminogen activator (tPA) mediated by N-methyl-d-aspartate (NMDA) receptors being toxic to neurons and inflicting damage to the neurovascular unit/blood–brain barrier (BBB) or regulating it in a way that leads to pathological consequences.
Figures
References
-
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013; 14: 383–400. - PubMed
-
- Triller A, Choquet D. Surface trafficking of receptors between synaptic and extrasynaptic membranes: and yet they do move!. Trends Neurosci 2005; 28: 133–139. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
