

Mitochondrial Genome Sequences and Structures Aid in the Resolution of Piroplasmida phylogeny
- PMID: 27832128
- PMCID: PMC5104439
- DOI: 10.1371/journal.pone.0165702
Mitochondrial Genome Sequences and Structures Aid in the Resolution of Piroplasmida phylogeny
Abstract
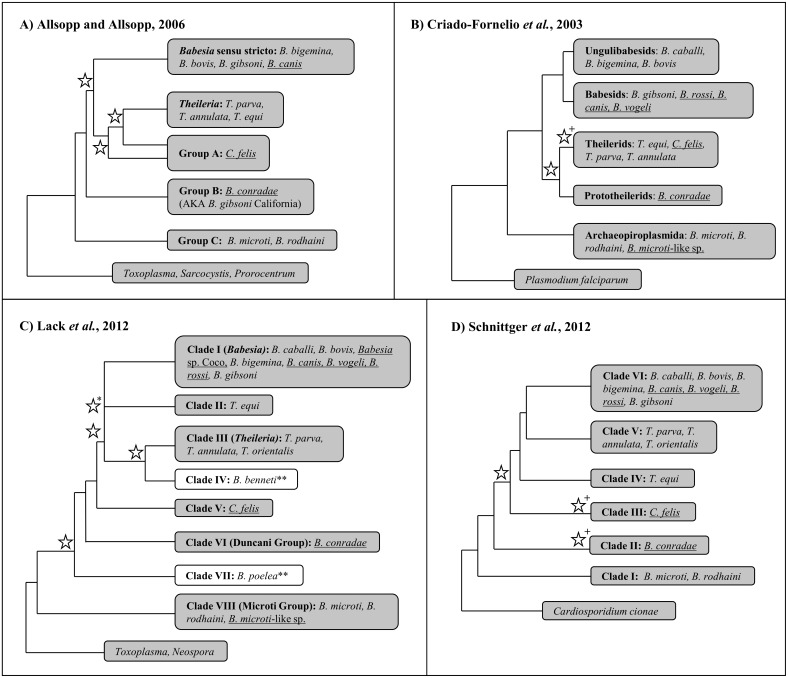
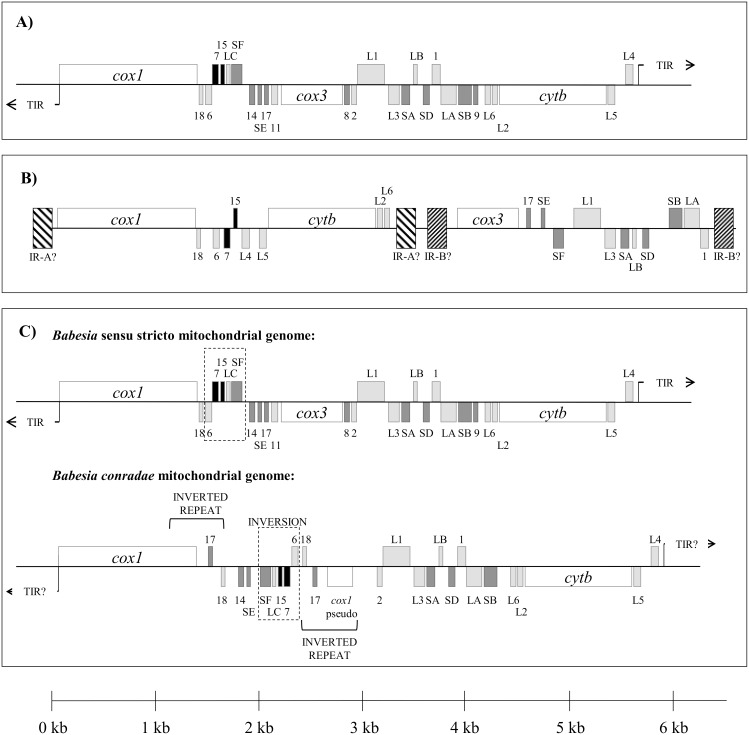
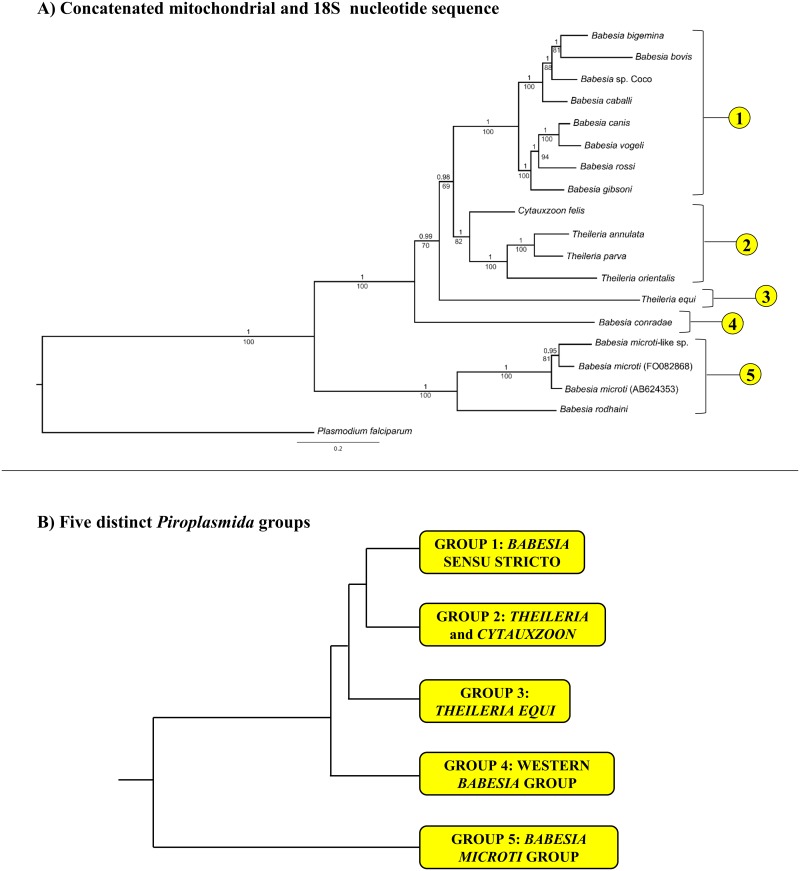
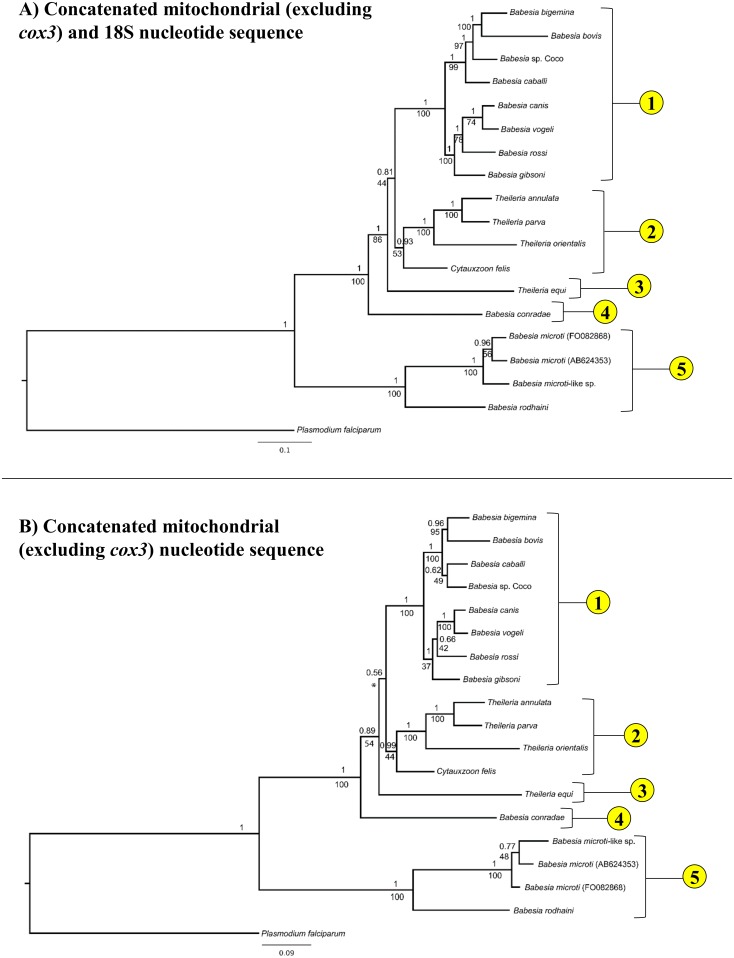
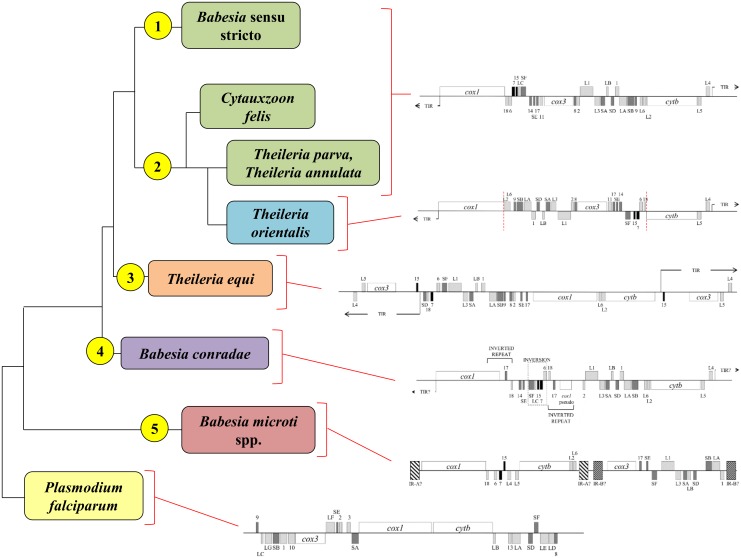
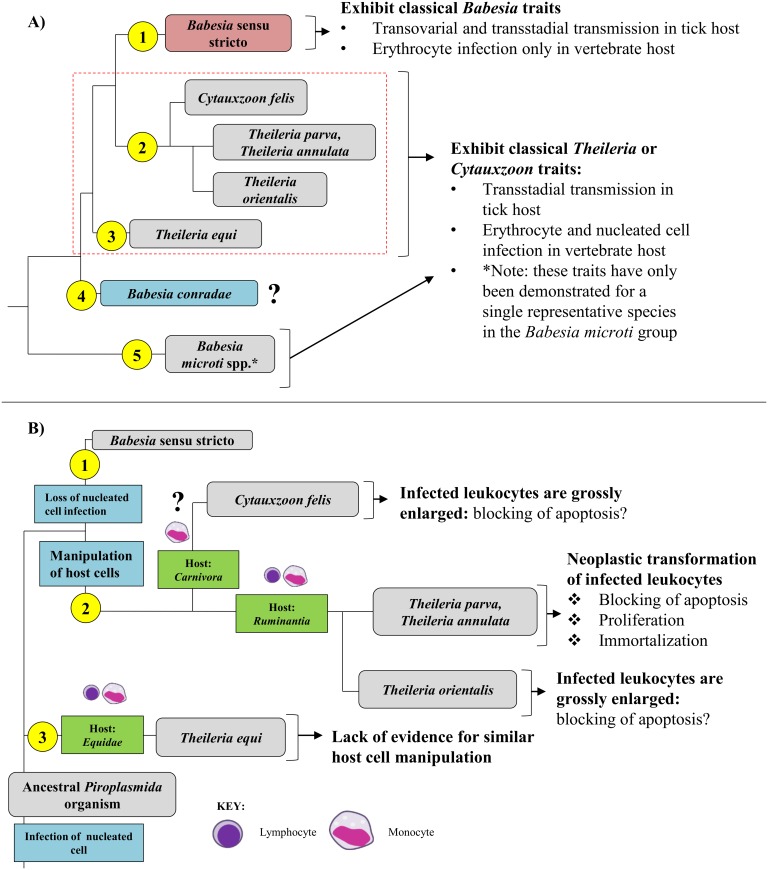
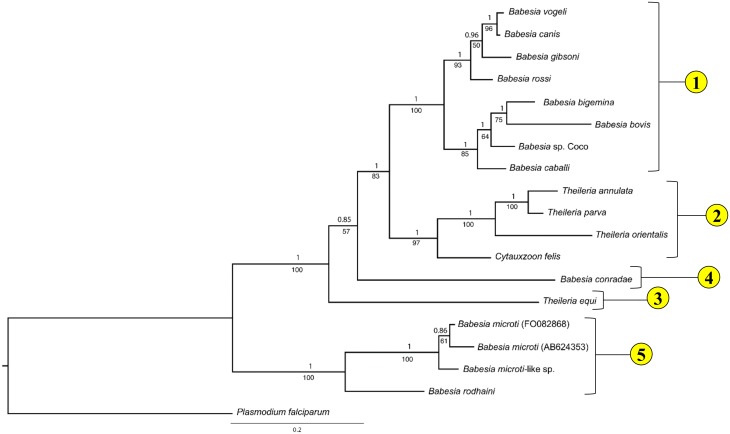
The taxonomy of the order Piroplasmida, which includes a number of clinically and economically relevant organisms, is a hotly debated topic amongst parasitologists. Three genera (Babesia, Theileria, and Cytauxzoon) are recognized based on parasite life cycle characteristics, but molecular phylogenetic analyses of 18S sequences have suggested the presence of five or more distinct Piroplasmida lineages. Despite these important advancements, a few studies have been unable to define the taxonomic relationships of some organisms (e.g. C. felis and T. equi) with respect to other Piroplasmida. Additional evidence from mitochondrial genome sequences and synteny should aid in the inference of Piroplasmida phylogeny and resolution of taxonomic uncertainties. In this study, we have amplified, sequenced, and annotated seven previously uncharacterized mitochondrial genomes (Babesia canis, Babesia vogeli, Babesia rossi, Babesia sp. Coco, Babesia conradae, Babesia microti-like sp., and Cytauxzoon felis) and identified additional ribosomal fragments in ten previously characterized mitochondrial genomes. Phylogenetic analysis of concatenated mitochondrial and 18S sequences as well as cox1 amino acid sequence identified five distinct Piroplasmida groups, each of which possesses a unique mitochondrial genome structure. Specifically, our results confirm the existence of four previously identified clades (B. microti group, Babesia sensu stricto, Theileria equi, and a Babesia sensu latu group that includes B. conradae) while supporting the integration of Theileria and Cytauxzoon species into a single fifth taxon. Although known biological characteristics of Piroplasmida corroborate the proposed phylogeny, more investigation into parasite life cycles is warranted to further understand the evolution of the Piroplasmida. Our results provide an evolutionary framework for comparative biology of these important animal and human pathogens and help focus renewed efforts toward understanding the phylogenetic relationships within the group.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Bishop R, Musoke A, Morzaria S, Gardner M, Nene V (2004) Theileria: intracellular protozoan parasites of wild and domestic ruminants transmitted by ixodid ticks. Parasitology 129 Suppl: S271–283. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
