

Exome sequencing identifies pathogenic variants of VPS13B in a patient with familial 16p11.2 duplication
- PMID: 27832746
- PMCID: PMC5105257
- DOI: 10.1186/s12881-016-0340-0
Exome sequencing identifies pathogenic variants of VPS13B in a patient with familial 16p11.2 duplication
Abstract
Background: The recurrent microduplication of 16p11.2 (dup16p11.2) is associated with a broad spectrum of neurodevelopmental disorders (NDD) confounded by incomplete penetrance and variable expressivity. This inter- and intra-familial clinical variability highlights the importance of personalized genetic counselling in individuals at-risk.
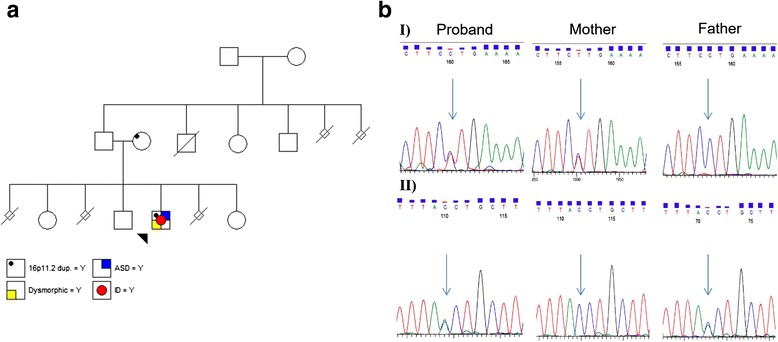
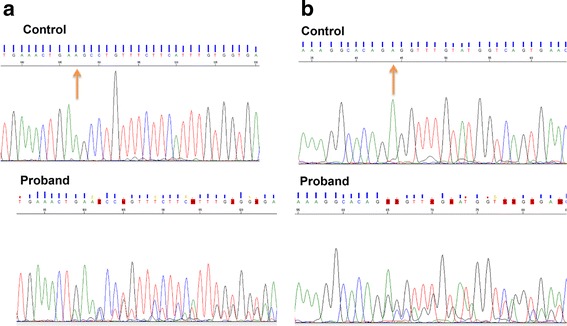
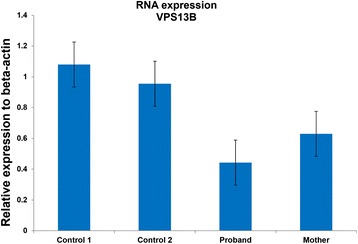
Case presentation: In this study, we performed whole exome sequencing (WES) to look for other genomic alterations that could explain the clinical variability in a family with a boy presenting with NDD who inherited the dup16p11.2 from his apparently healthy mother. We identified novel splicing variants of VPS13B (8q22.2) in the proband with compound heterozygous inheritance. Two VPS13B mutations abolished the canonical splice sites resulting in low RNA expression in transformed lymphoblasts of the proband. VPS13B mutation causes Cohen syndrome (CS) consistent with the proband's phenotype (intellectual disability (ID), microcephaly, facial gestalt, retinal dystrophy, joint hypermobility and neutropenia). The new diagnosis of CS has important health implication for the proband, provides the opportunity for more meaningful and accurate genetic counselling for the family; and underscores the importance of longitudinally following patients for evolving phenotypic features.
Conclusions: This is the first report of a co-occurrence of pathogenic variants with familial dup16p11.2. Our finding suggests that the variable expressivity among carriers of rare putatively pathogenic CNVs such as dup16p11.2 warrants further study by WES and individualized genetic counselling of families with such CNVs.
Keywords: 16p11.2 duplication; Case report; Cohen syndrome; Neuro-developmental disorders; Variable expressivity; Whole exome sequencing.
Figures
Similar articles
-
Value of whole exome sequencing for syndromic retinal dystrophy diagnosis in young patients.Clin Exp Ophthalmol. 2015 Mar;43(2):132-8. doi: 10.1111/ceo.12391. Epub 2014 Oct 2. Clin Exp Ophthalmol. 2015. PMID: 25060287
-
CNV analysis using whole exome sequencing identified biallelic CNVs of VPS13B in siblings with intellectual disability.Eur J Med Genet. 2020 Jan;63(1):103610. doi: 10.1016/j.ejmg.2018.12.015. Epub 2018 Dec 30. Eur J Med Genet. 2020. PMID: 30602132
-
Homozygosity mapping and whole exome sequencing provide exact diagnosis of Cohen syndrome in a Saudi family.Brain Dev. 2020 Sep;42(8):587-593. doi: 10.1016/j.braindev.2020.04.010. Epub 2020 May 10. Brain Dev. 2020. PMID: 32402540
-
A novel VPS13B mutation in Cohen syndrome: a case report and review of literature.BMC Med Genet. 2020 Jun 30;21(1):140. doi: 10.1186/s12881-020-01075-1. BMC Med Genet. 2020. PMID: 32605629 Free PMC article. Review.
-
Novel Compound Heterozygous Variants in ZNF526 Causing Dentici-Novelli Neurodevelopmental Syndrome: A Case Report and Literature Review.Mol Genet Genomic Med. 2025 Apr;13(4):e70089. doi: 10.1002/mgg3.70089. Mol Genet Genomic Med. 2025. PMID: 40197775 Free PMC article. Review.
Cited by
-
Case Report: Prenatal Whole-Exome Sequencing Identified a Novel Nonsense Mutation of the KCNH2 Gene in a Fetus With Familial 2q14.2 Duplication.Front Genet. 2022 Jul 5;13:924573. doi: 10.3389/fgene.2022.924573. eCollection 2022. Front Genet. 2022. PMID: 35865016 Free PMC article.
-
The Role of Visual Electrophysiology in Systemic Hereditary Syndromes.Int J Mol Sci. 2025 Jan 23;26(3):957. doi: 10.3390/ijms26030957. Int J Mol Sci. 2025. PMID: 39940729 Free PMC article. Review.
-
Molecular cytogenetic characterization of isolated recurrent 4q35.2 microduplication in Chinese population: a seven-year single-center retrospective study.BMC Pregnancy Childbirth. 2024 Sep 18;24(1):606. doi: 10.1186/s12884-024-06818-z. BMC Pregnancy Childbirth. 2024. PMID: 39294589 Free PMC article.
-
Prenatal diagnosis of fetuses with 15q11.2 BP1-BP2 microdeletion in the Chinese population: a seven-year single-center retrospective study.Mol Cytogenet. 2024 Sep 2;17(1):20. doi: 10.1186/s13039-024-00690-4. Mol Cytogenet. 2024. PMID: 39218907 Free PMC article.
-
A Novel Homozygous VPS13B Splice-Site Mutation Causing the Skipping of Exon 38 in a Chinese Family With Cohen Syndrome.Front Pediatr. 2021 Apr 20;9:651621. doi: 10.3389/fped.2021.651621. eCollection 2021. Front Pediatr. 2021. PMID: 33959574 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
