

Molecular inotropy mediated by cardiac miR-based PDE4D/PRKAR1α/phosphoprotein signaling
- PMID: 27833092
- PMCID: PMC5105063
- DOI: 10.1038/srep36803
Molecular inotropy mediated by cardiac miR-based PDE4D/PRKAR1α/phosphoprotein signaling
Abstract
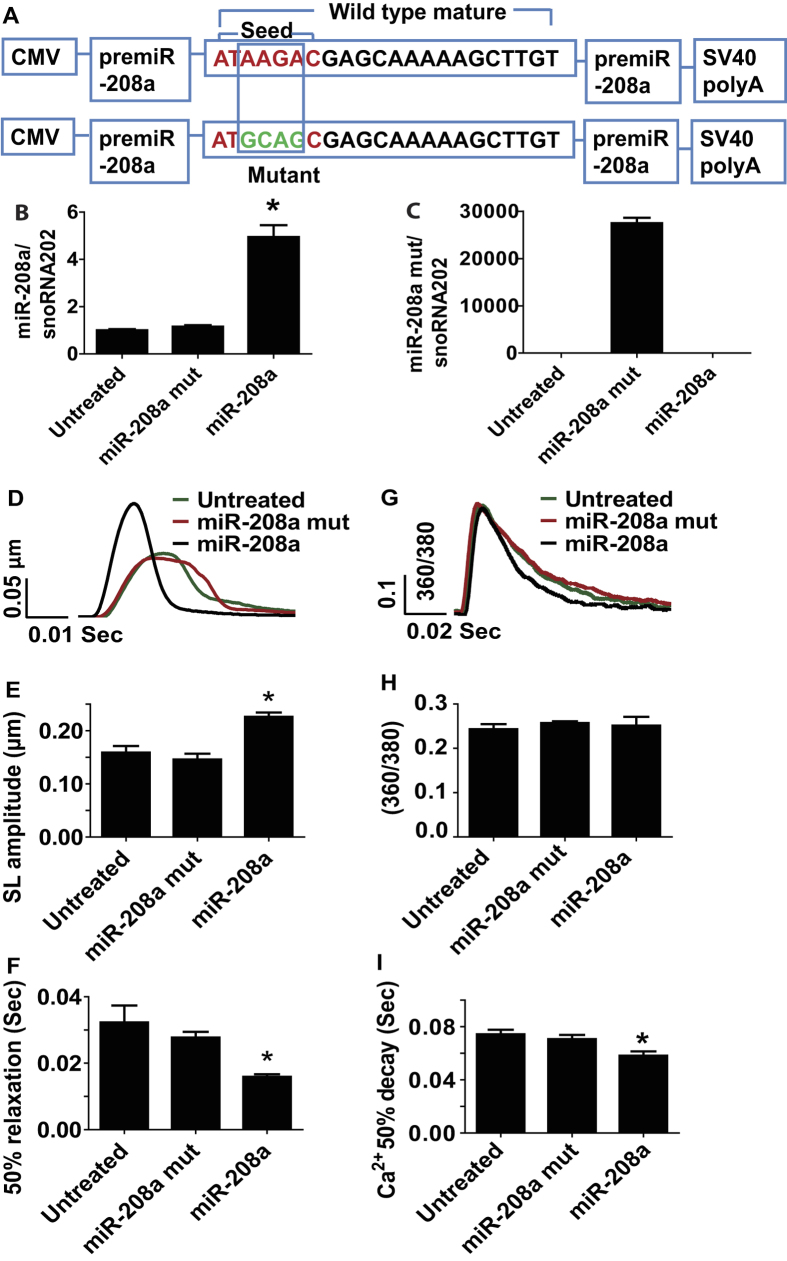
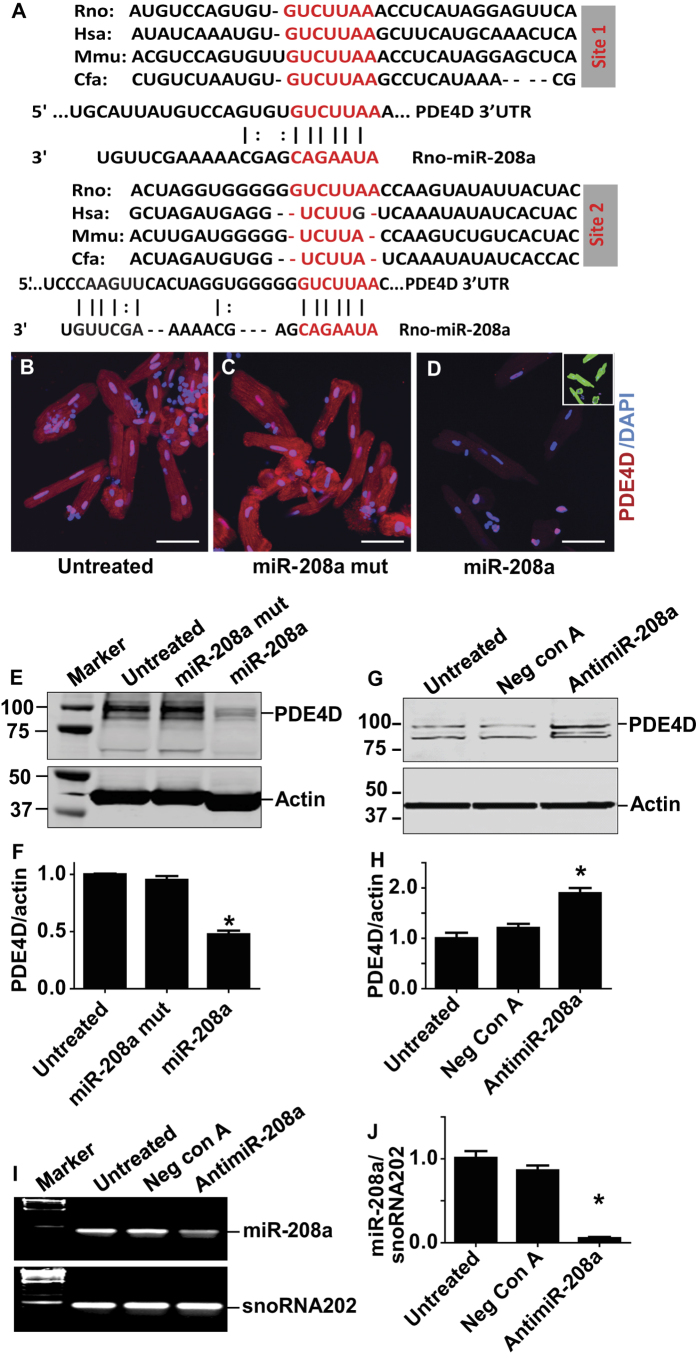
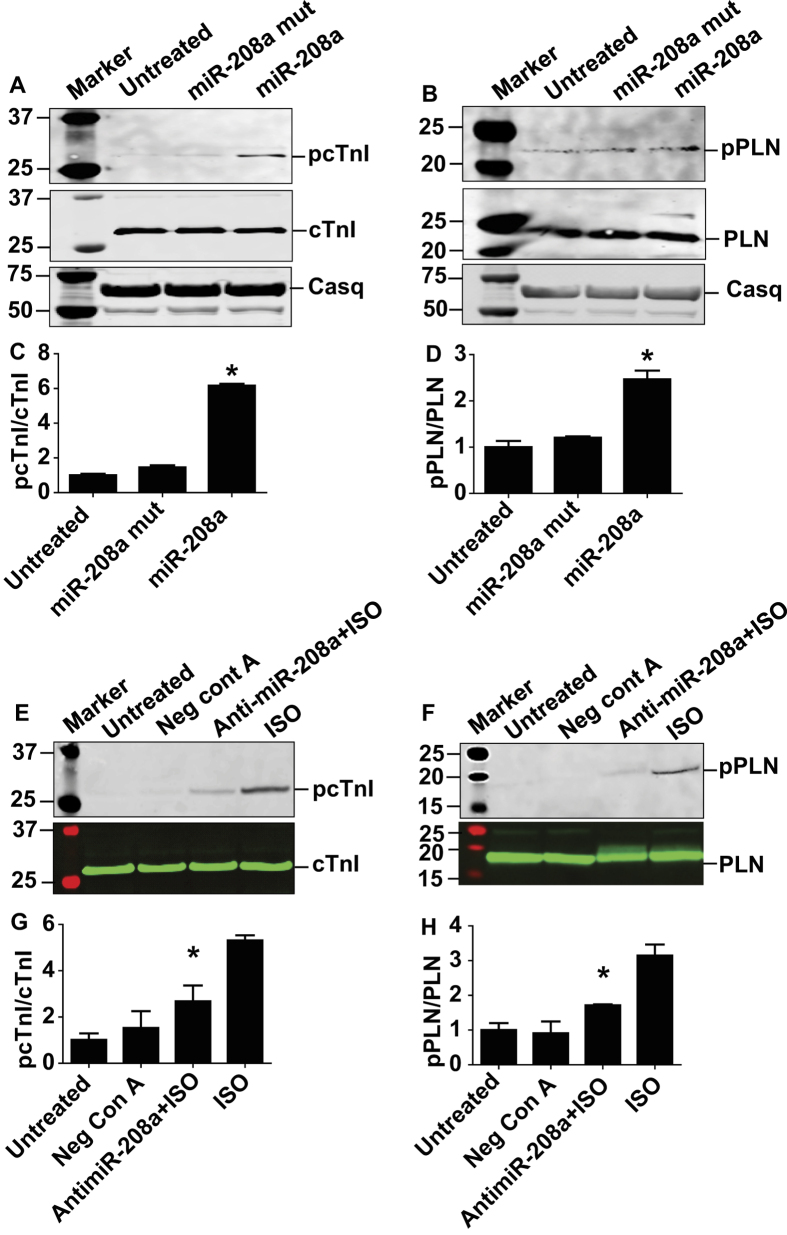
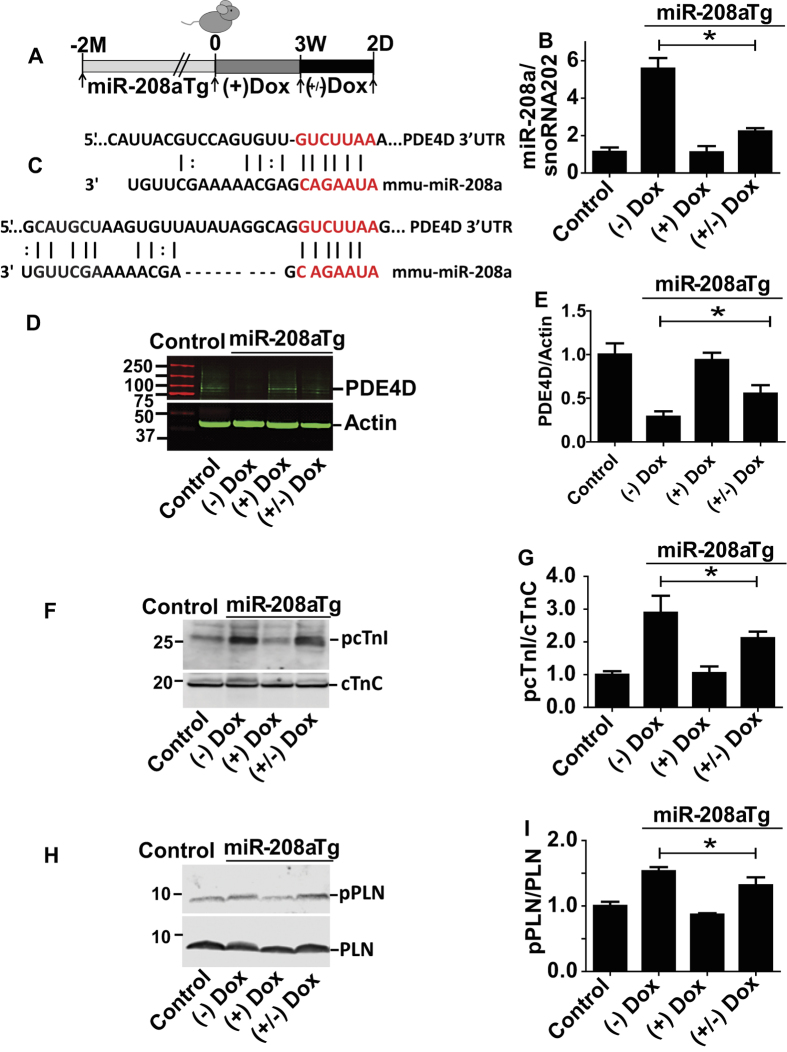
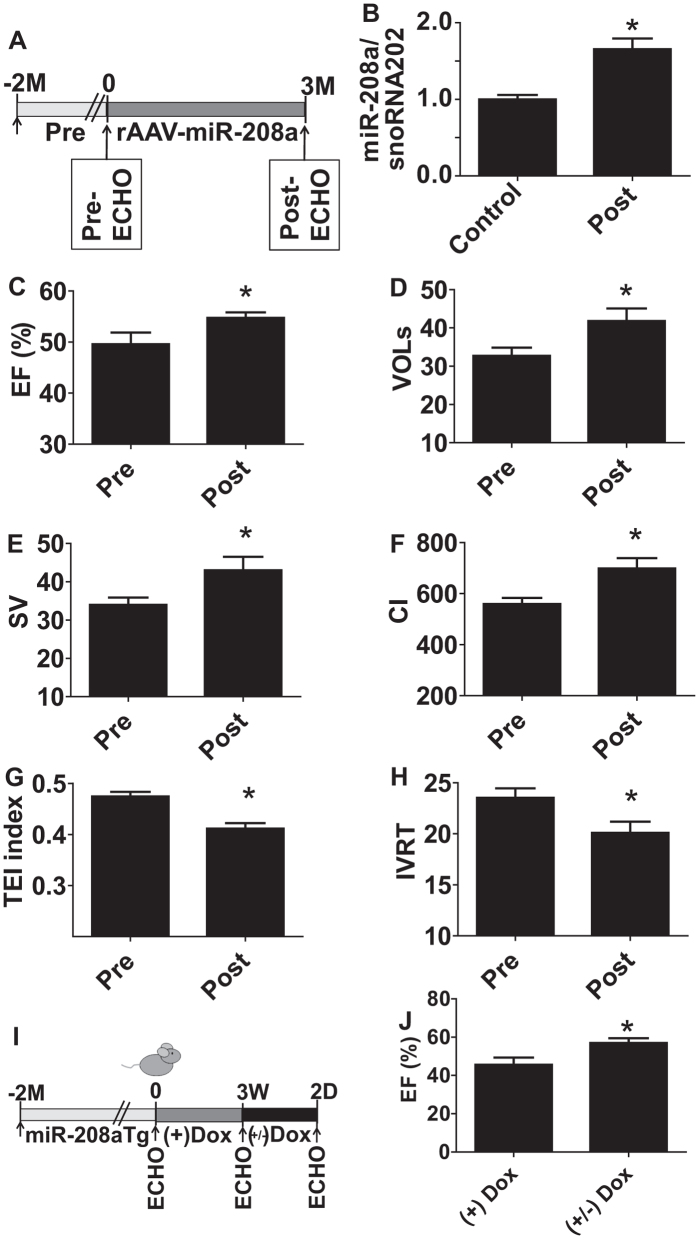
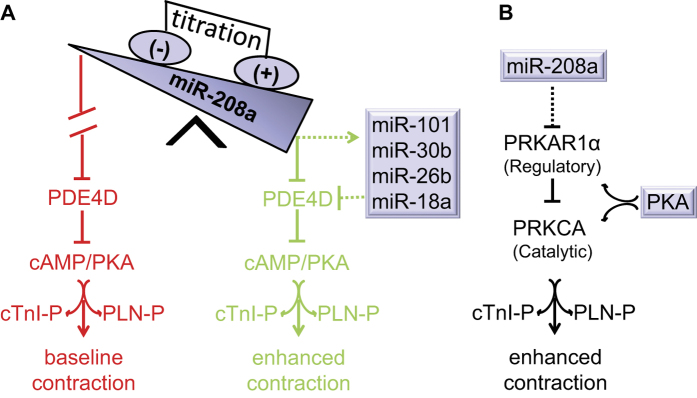
Molecular inotropy refers to cardiac contractility that can be modified to affect overall heart pump performance. Here we show evidence of a new molecular pathway for positive inotropy by a cardiac-restricted microRNA (miR). We report enhanced cardiac myocyte performance by acute titration of cardiac myosin-embedded miR-208a. The observed positive effect was independent of host gene myosin effects with evidence of negative regulation of cAMP-specific 3',5'-cyclic phosphodiesterase 4D (PDE4D) and the regulatory subunit of PKA (PRKAR1α) content culminating in PKA-site dependent phosphorylation of cardiac troponin I (cTnI) and phospholamban (PLN). Further, acute inhibition of miR-208a in adult myocytes in vitro increased PDE4D expression causing reduced isoproterenol-mediated phosphorylation of cTnI and PLN. Next, rAAV-mediated miR-208a gene delivery enhanced heart contractility and relaxation parameters in vivo. Finally, acute inducible increases in cardiac miR-208a in vivo reduced PDE4D and PRKAR1α, with evidence of increased content of several complementary miRs harboring the PDE4D recognition sequence. Physiologically, this resulted in significant cardiac cTnI and PLN phosphorylation and improved heart performance in vivo. As phosphorylation of cTnI and PLN is critical to myocyte function, titration of miR-208a represents a potential new mechanism to enhance myocardial performance via the PDE4D/PRKAR1α/PKA phosphoprotein signaling pathway.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
