

Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study
- PMID: 27834756
- PMCID: PMC5502308
- DOI: 10.1136/jmedgenet-2016-104178
Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study
Erratum in
-
Correction: Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study.J Med Genet. 2018 Jun;55(6):422-429. doi: 10.1136/jmedgenet-2016-104178corr1. Epub 2018 Apr 16. J Med Genet. 2018. PMID: 29661968 Free PMC article. No abstract available.
Abstract
Background: Fabry disease is an X-linked lysosomal storage disorder caused by GLA mutations, resulting in α-galactosidase (α-Gal) deficiency and accumulation of lysosomal substrates. Migalastat, an oral pharmacological chaperone being developed as an alternative to intravenous enzyme replacement therapy (ERT), stabilises specific mutant (amenable) forms of α-Gal to facilitate normal lysosomal trafficking.
Methods: The main objective of the 18-month, randomised, active-controlled ATTRACT study was to assess the effects of migalastat on renal function in patients with Fabry disease previously treated with ERT. Effects on heart, disease substrate, patient-reported outcomes (PROs) and safety were also assessed.
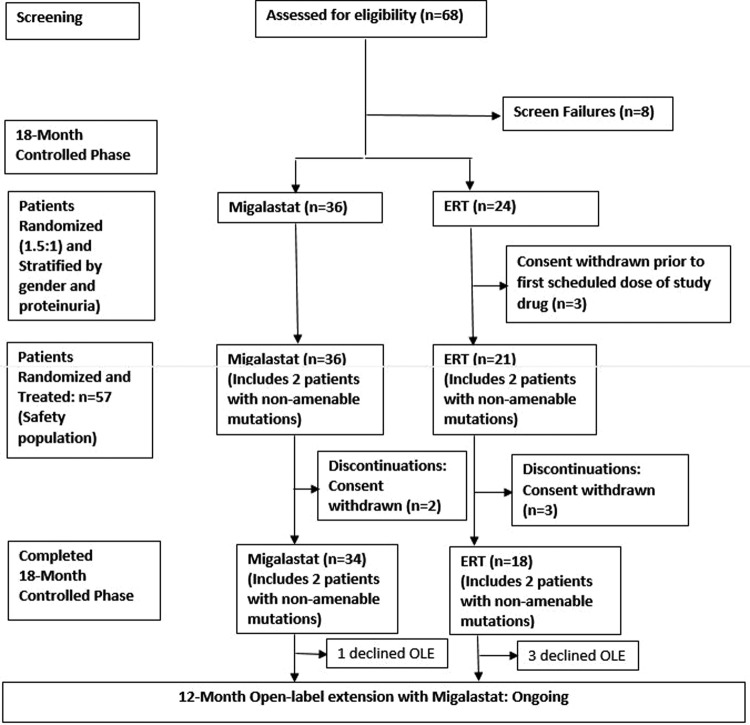
Results: Fifty-seven adults (56% female) receiving ERT (88% had multiorgan disease) were randomised (1.5:1), based on a preliminary cell-based assay of responsiveness to migalastat, to receive 18 months open-label migalastat or remain on ERT. Four patients had non-amenable mutant forms of α-Gal based on the validated cell-based assay conducted after treatment initiation and were excluded from primary efficacy analyses only. Migalastat and ERT had similar effects on renal function. Left ventricular mass index decreased significantly with migalastat treatment (-6.6 g/m2 (-11.0 to -2.2)); there was no significant change with ERT. Predefined renal, cardiac or cerebrovascular events occurred in 29% and 44% of patients in the migalastat and ERT groups, respectively. Plasma globotriaosylsphingosine remained low and stable following the switch from ERT to migalastat. PROs were comparable between groups. Migalastat was generally safe and well tolerated.
Conclusions: Migalastat offers promise as a first-in-class oral monotherapy alternative treatment to intravenous ERT for patients with Fabry disease and amenable mutations.
Trial registration number: NCT00925301; Pre-results.
Trial registration: ClinicalTrials.gov NCT01218659.
Keywords: Fabry disease; Pharmacological chaperone; enzyme replacement therapy; lyso-Gb3; lysosomal storage disorder.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/.
Conflict of interest statement
Figures
References
-
- Khanna R, Soska R, Lun Y, Feng J, Frascella M, Young B, Brignol N, Pellegrino L, Sitaraman SA, Desnick RJ, Benjamin ER, Lockhart DJ, Valenzano KJ. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol Ther 2010;18:23–33. 10.1038/mt.2009.220 - DOI - PMC - PubMed
-
- Benjamin ER, Flanagan JJ, Schilling A, Chang HH, Agarwal L, Katz E, Wu X, Pine C, Wustman B, Desnick RJ, Lockhart DJ, Valenzano KJ. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J Inherit Metab Dis 2009;32:424–40. 10.1007/s10545-009-1077-0 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous