

A MAD-Bayes Algorithm for State-Space Inference and Clustering with Application to Querying Large Collections of ChIP-Seq Data Sets
- PMID: 27835030
- PMCID: PMC5467113
- DOI: 10.1089/cmb.2016.0138
A MAD-Bayes Algorithm for State-Space Inference and Clustering with Application to Querying Large Collections of ChIP-Seq Data Sets
Abstract
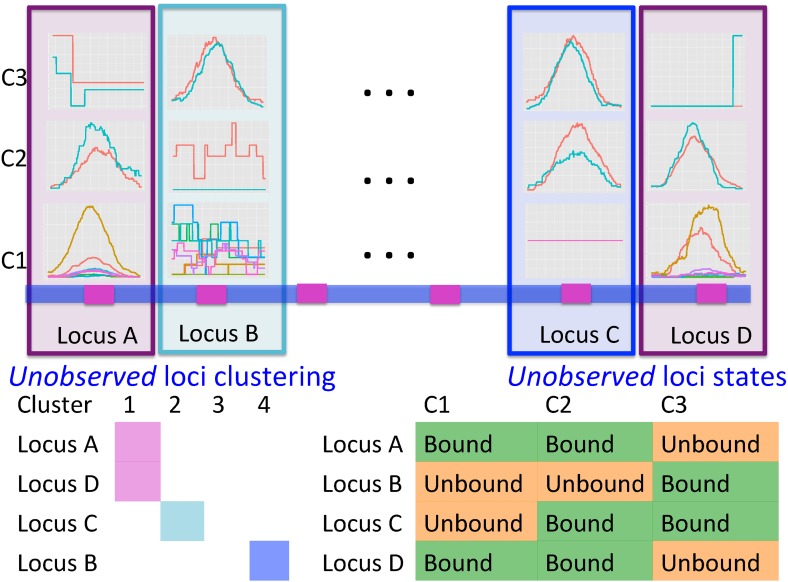
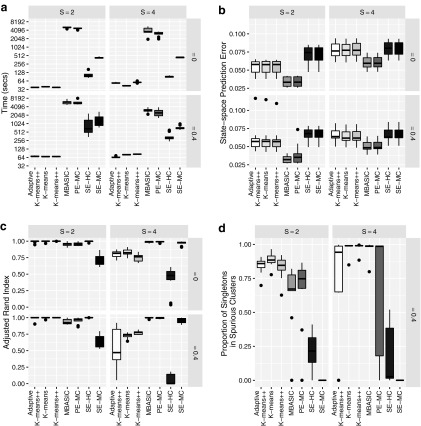
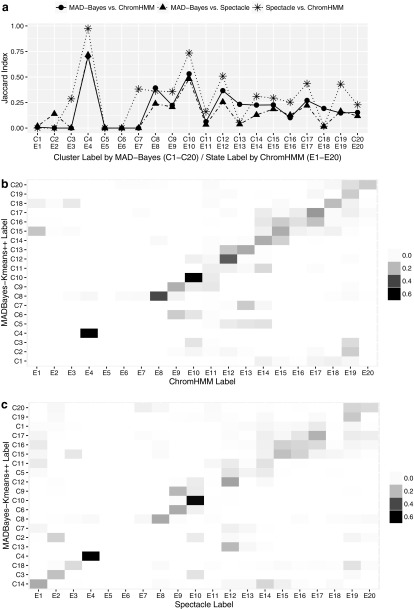
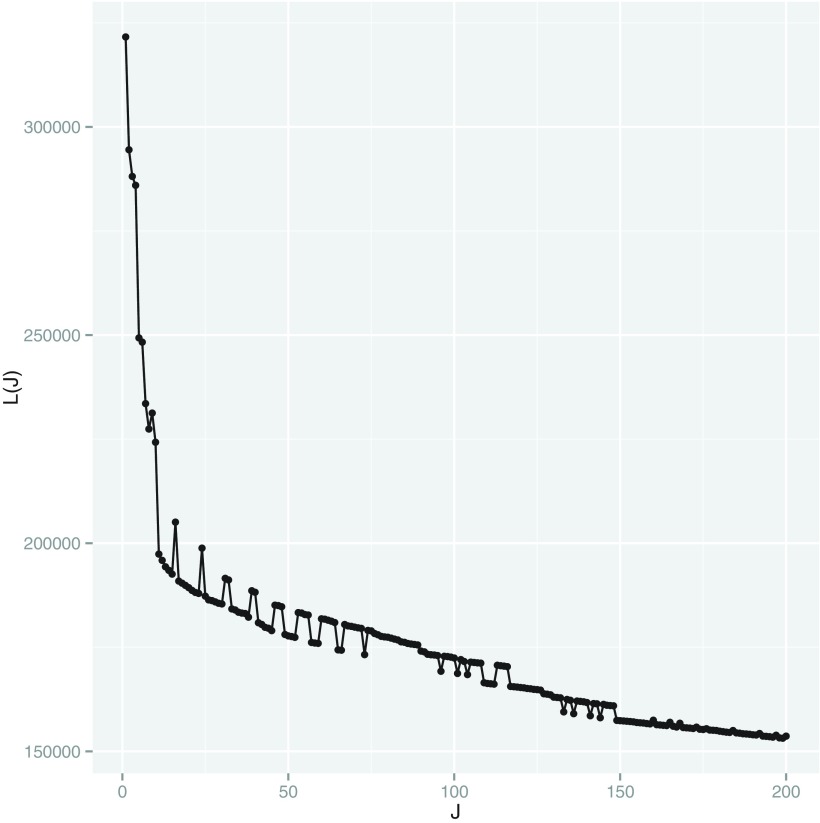
Current analytic approaches for querying large collections of chromatin immunoprecipitation followed by sequencing (ChIP-seq) data from multiple cell types rely on individual analysis of each data set (i.e., peak calling) independently. This approach discards the fact that functional elements are frequently shared among related cell types and leads to overestimation of the extent of divergence between different ChIP-seq samples. Methods geared toward multisample investigations have limited applicability in settings that aim to integrate 100s to 1000s of ChIP-seq data sets for query loci (e.g., thousands of genomic loci with a specific binding site). Recently, Zuo et al. developed a hierarchical framework for state-space matrix inference and clustering, named MBASIC, to enable joint analysis of user-specified loci across multiple ChIP-seq data sets. Although this versatile framework estimates both the underlying state-space (e.g., bound vs. unbound) and also groups loci with similar patterns together, its Expectation-Maximization-based estimation structure hinders its applicability with large number of loci and samples. We address this limitation by developing MAP-based asymptotic derivations from Bayes (MAD-Bayes) framework for MBASIC. This results in a K-means-like optimization algorithm that converges rapidly and hence enables exploring multiple initialization schemes and flexibility in tuning. Comparison with MBASIC indicates that this speed comes at a relatively insignificant loss in estimation accuracy. Although MAD-Bayes MBASIC is specifically designed for the analysis of user-specified loci, it is able to capture overall patterns of histone marks from multiple ChIP-seq data sets similar to those identified by genome-wide segmentation methods such as ChromHMM and Spectacle.
Keywords: ChIP-Seq; MAD-Bayes; small-variance asymptotics; unified state-space inference and clustering.
Conflict of interest statement
No competing financial interests exist.
Figures

Similar articles
-
Software for rapid time dependent ChIP-sequencing analysis (TDCA).BMC Bioinformatics. 2017 Nov 25;18(1):521. doi: 10.1186/s12859-017-1936-x. BMC Bioinformatics. 2017. PMID: 29178831 Free PMC article.
-
Comparing genome-wide chromatin profiles using ChIP-chip or ChIP-seq.Bioinformatics. 2010 Apr 15;26(8):1000-6. doi: 10.1093/bioinformatics/btq087. Epub 2010 Mar 5. Bioinformatics. 2010. PMID: 20208068
-
Unified Analysis of Multiple ChIP-Seq Datasets.Methods Mol Biol. 2021;2198:451-465. doi: 10.1007/978-1-0716-0876-0_33. Methods Mol Biol. 2021. PMID: 32822050
-
Integrating ChIP-seq with other functional genomics data.Brief Funct Genomics. 2018 Mar 1;17(2):104-115. doi: 10.1093/bfgp/ely002. Brief Funct Genomics. 2018. PMID: 29579165 Free PMC article. Review.
-
[Processing and analysis of ChIP-seq data].Yi Chuan. 2012 Jun;34(6):773-83. doi: 10.3724/sp.j.1005.2012.00773. Yi Chuan. 2012. PMID: 22698750 Review. Chinese.
References
-
- Aldous D.J. 1983. Exchangeability and related topics, 1–198. In École d'Été de Probabilités de Saint-Flour XIII 1983. Ed: Hennequin P.L. Springer, Berlin; Heidelberg
-
- Banerjee A. 2005. Clustering with Bregman divergences. J. Mach. Learn. Res. 6, 1705–1749
-
- Bao Y., Vinciotti V., Wit E., et al. . 2014. Joint modeling of ChIP-seq data via a Markov random field model. Biostatistics 15, 296–310 - PubMed
-
- Bardet A.F., He Q., Zeitlinger J., and Stark A. 2012. A computational pipeline for comparative chip-seq analyses. Nat. Protoc. 7, 45–61 - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
