

Survey of the Heritability and Sparse Architecture of Gene Expression Traits across Human Tissues
- PMID: 27835642
- PMCID: PMC5106030
- DOI: 10.1371/journal.pgen.1006423
Survey of the Heritability and Sparse Architecture of Gene Expression Traits across Human Tissues
Abstract
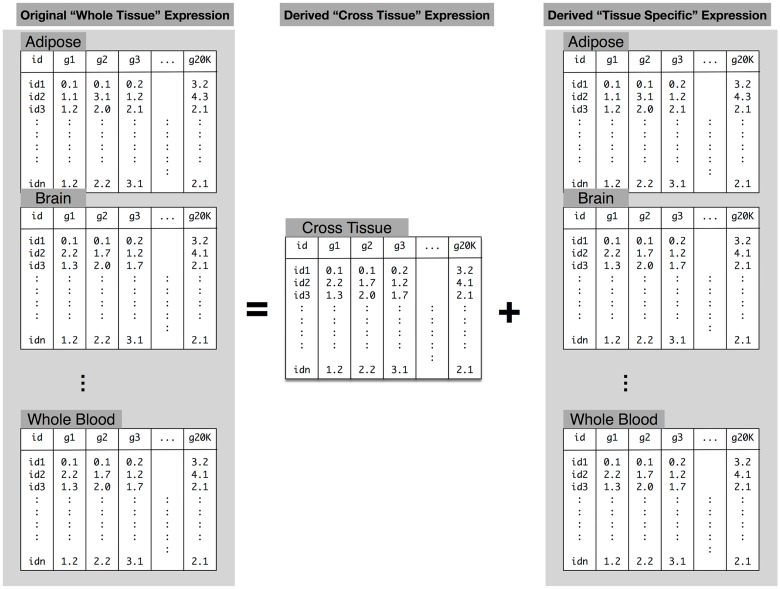
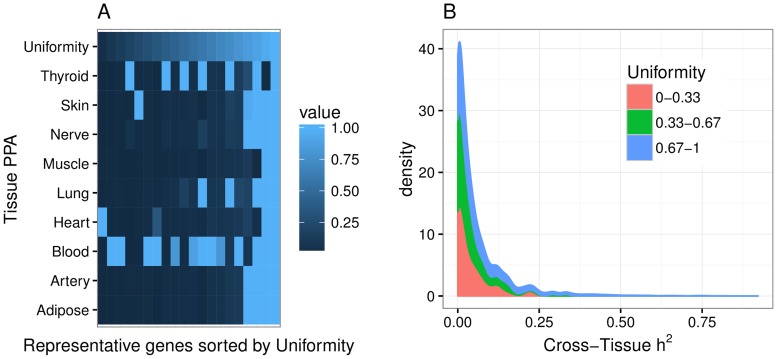
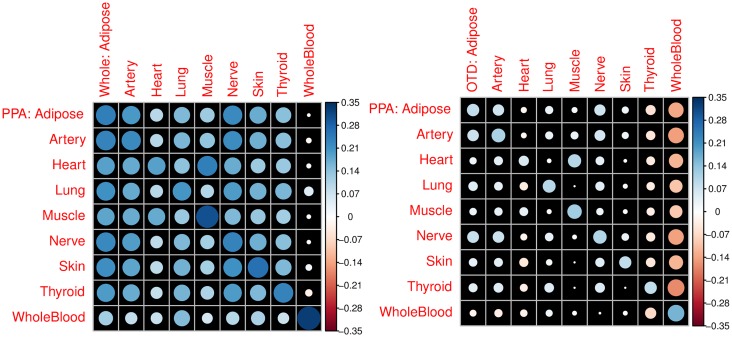
Understanding the genetic architecture of gene expression traits is key to elucidating the underlying mechanisms of complex traits. Here, for the first time, we perform a systematic survey of the heritability and the distribution of effect sizes across all representative tissues in the human body. We find that local h2 can be relatively well characterized with 59% of expressed genes showing significant h2 (FDR < 0.1) in the DGN whole blood cohort. However, current sample sizes (n ≤ 922) do not allow us to compute distal h2. Bayesian Sparse Linear Mixed Model (BSLMM) analysis provides strong evidence that the genetic contribution to local expression traits is dominated by a handful of genetic variants rather than by the collective contribution of a large number of variants each of modest size. In other words, the local architecture of gene expression traits is sparse rather than polygenic across all 40 tissues (from DGN and GTEx) examined. This result is confirmed by the sparsity of optimal performing gene expression predictors via elastic net modeling. To further explore the tissue context specificity, we decompose the expression traits into cross-tissue and tissue-specific components using a novel Orthogonal Tissue Decomposition (OTD) approach. Through a series of simulations we show that the cross-tissue and tissue-specific components are identifiable via OTD. Heritability and sparsity estimates of these derived expression phenotypes show similar characteristics to the original traits. Consistent properties relative to prior GTEx multi-tissue analysis results suggest that these traits reflect the expected biology. Finally, we apply this knowledge to develop prediction models of gene expression traits for all tissues. The prediction models, heritability, and prediction performance R2 for original and decomposed expression phenotypes are made publicly available (https://github.com/hakyimlab/PrediXcan).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Grants and funding
- R01 MH101782/MH/NIMH NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- N01 HC025195/HL/NHLBI NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- R01 MH101822/MH/NIMH NIH HHS/United States
- P60 DK020595/DK/NIDDK NIH HHS/United States
- K12 CA139160/CA/NCI NIH HHS/United States
- R01 DA006227/DA/NIDA NIH HHS/United States
- HHSN268201500001C/HL/NHLBI NIH HHS/United States
- RC2 MH089916/MH/NIMH NIH HHS/United States
- R01 MH107666/MH/NIMH NIH HHS/United States
- R01 MH101810/MH/NIMH NIH HHS/United States
- P50 MH094267/MH/NIMH NIH HHS/United States
- HHSN268201500001I/HL/NHLBI NIH HHS/United States
- T32 MH020065/MH/NIMH NIH HHS/United States
- P50 DA037844/DA/NIDA NIH HHS/United States
- R01 MH101825/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
