

Reactive Oxygen Species/Nitric Oxide Mediated Inter-Organ Communication in Skeletal Muscle Wasting Diseases
- PMID: 27835923
- PMCID: PMC5421600
- DOI: 10.1089/ars.2016.6942
Reactive Oxygen Species/Nitric Oxide Mediated Inter-Organ Communication in Skeletal Muscle Wasting Diseases
Abstract
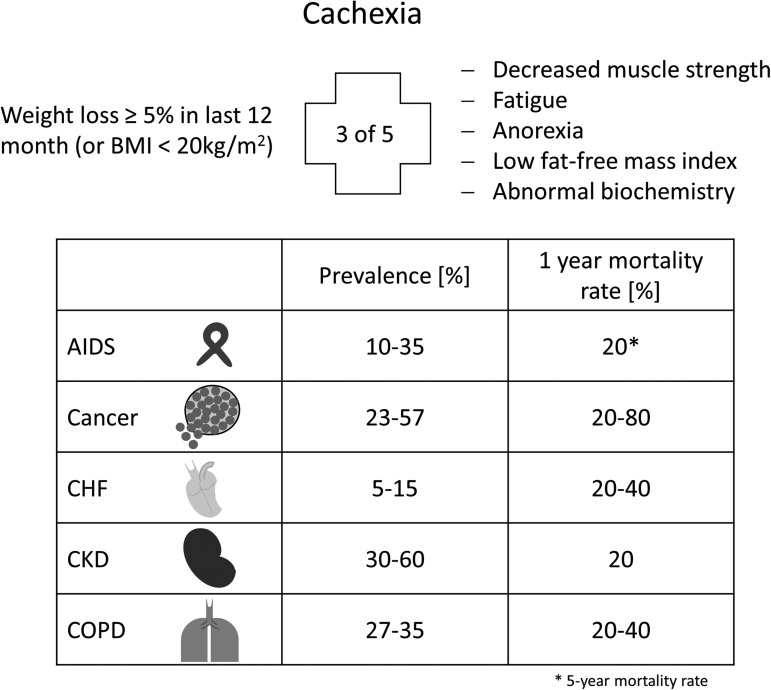
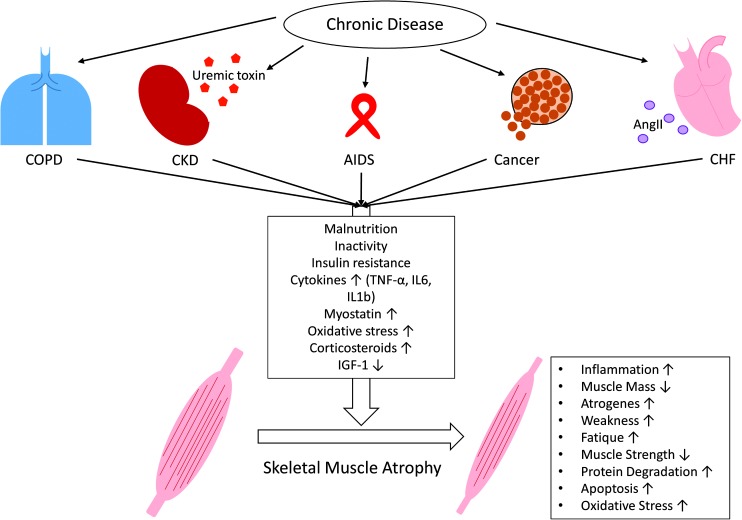
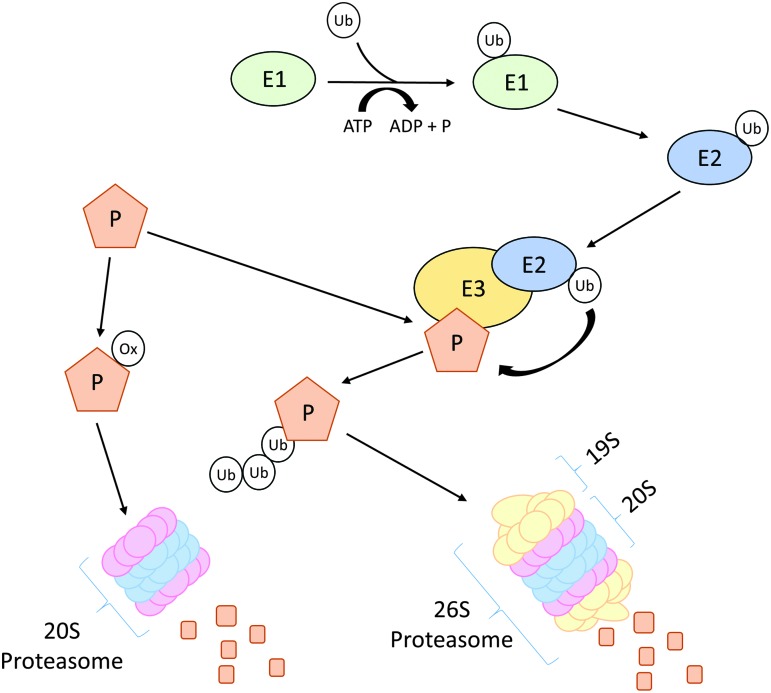
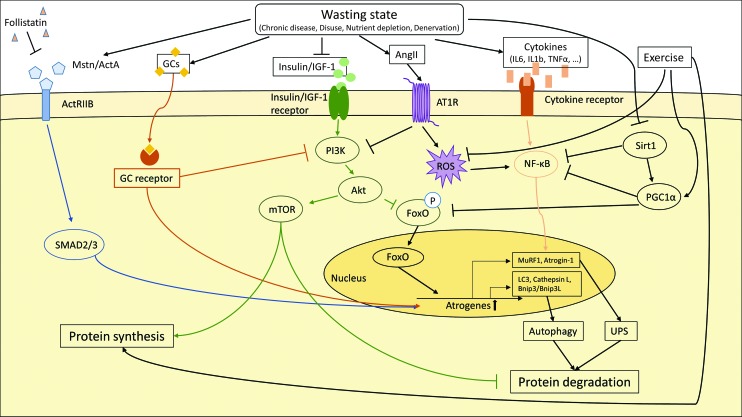
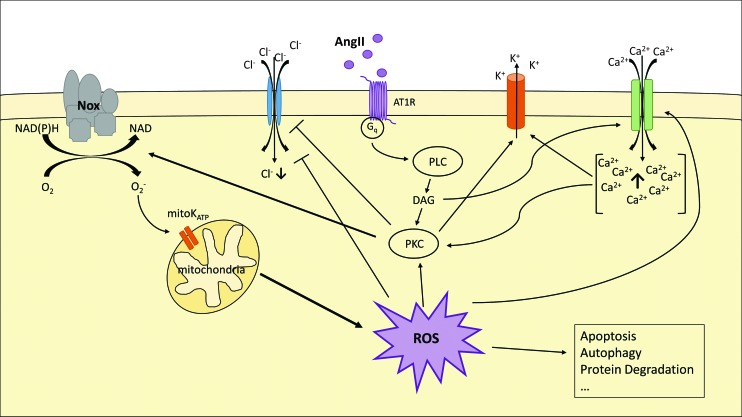
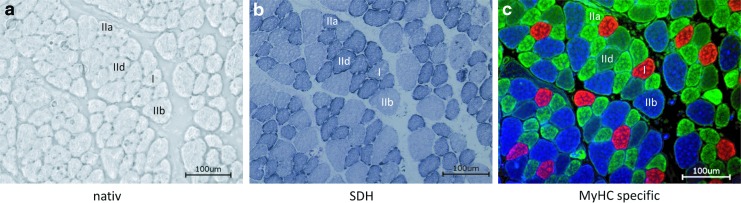
Significance: Cachexia is defined as a complex metabolic syndrome that is associated with underlying illness and a loss of muscle with or without loss of fat mass. This disease is associated with a high incidence with chronic diseases such as heart failure, cancer, chronic obstructive pulmonary disease (COPD), and acquired immunodeficiency syndrome (AIDS), among others. Since there is currently no effective treatment available, cachectic patients have a poor prognosis. Elucidation of the underlying mechanisms is, therefore, an important medical task. Recent Advances: There is accumulating evidence that the diseased organs such as heart, lung, kidney, or cancer tissue secrete soluble factors, including Angiotensin II, myostatin (growth differentiation factor 8 [GDF8]), GDF11, tumor growth factor beta (TGFβ), which act on skeletal muscle. There, they induce a set of genes called atrogenes, which, among others, induce the ubiquitin-proteasome system, leading to protein degradation. Moreover, elevated reactive oxygen species (ROS) levels due to modulation of NADPH oxidases (Nox) and mitochondrial function contribute to disease progression, which is characterized by loss of muscle mass, exercise resistance, and frailty.
Critical issues: Although substantial progress was achieved to elucidate the pathophysiology of cachexia, effectice therapeutic strategies are urgently needed.
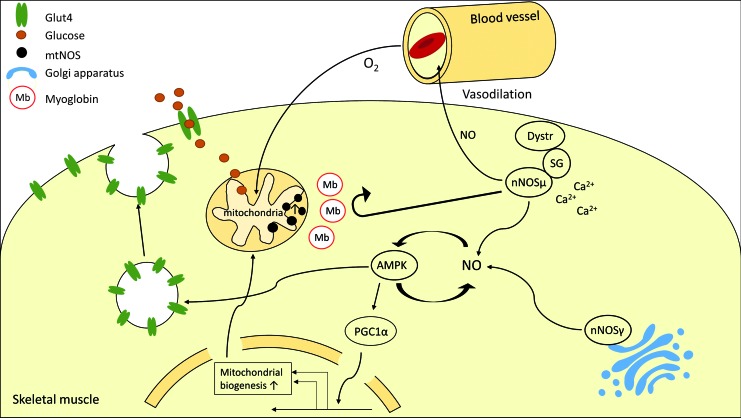
Future directions: With the identification of key components of the aberrant inter-organ communication leading to cachexia, studies in mice and men to inhibit ROS formation, induction of anti-oxidative superoxide dismutases, and upregulation of muscular nitric oxide (NO) formation either by pharmacological tools or by exercise are promising approaches to reduce the extent of skeletal muscle wasting. Antioxid. Redox Signal. 26, 700-717.
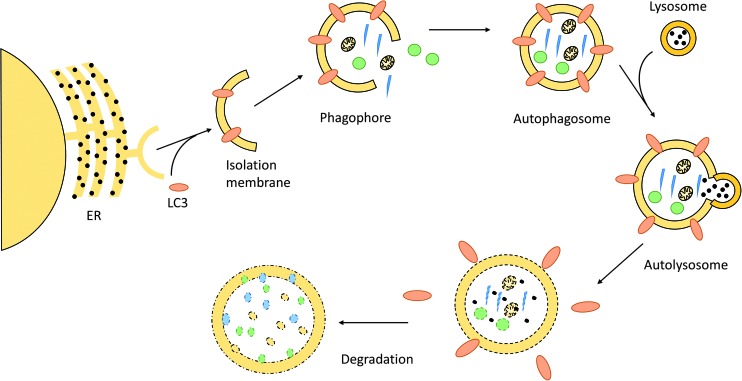
Keywords: angiotensin; autophagy; cachexia; exercise; skeletal muscle; ubiquitin.
Figures
References
-
- MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 360: 23–33, 2002 - PubMed
-
- Abrigo J, Rivera JC, Simon F, Cabrera D, and Cabello-Verrugio C. Transforming growth factor type beta (TGF-beta) requires reactive oxygen species to induce skeletal muscle atrophy. Cell Signal 28: 366–376, 2016 - PubMed
-
- Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, Ringel MD, Skipworth RJ, Fearon KC, Hollingsworth MA, Muscarella P, Burghes AH, Rafael-Fortney JA, and Guttridge DC. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 8: 421–432, 2005 - PubMed
-
- Adams V, Linke A, Krankel N, Erbs S, Gielen S, Mobius-Winkler S, Gummert JF, Mohr FW, Schuler G, and Hambrecht R. Impact of regular physical activity on the NAD(P)H oxidase and angiotensin receptor system in patients with coronary artery disease. Circulation 111: 555–562, 2005 - PubMed
-
- Adams V, Yu J, Mobius-Winkler S, Linke A, Weigl C, Hilbrich L, Schuler G, and Hambrecht R. Increased inducible nitric oxide synthase in skeletal muscle biopsies from patients with chronic heart failure. Biochem Mol Med 61: 152–160, 1997 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
