

Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53
- PMID: 27836911
- PMCID: PMC5287061
- DOI: 10.1101/cshperspect.a026054
Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53
Abstract
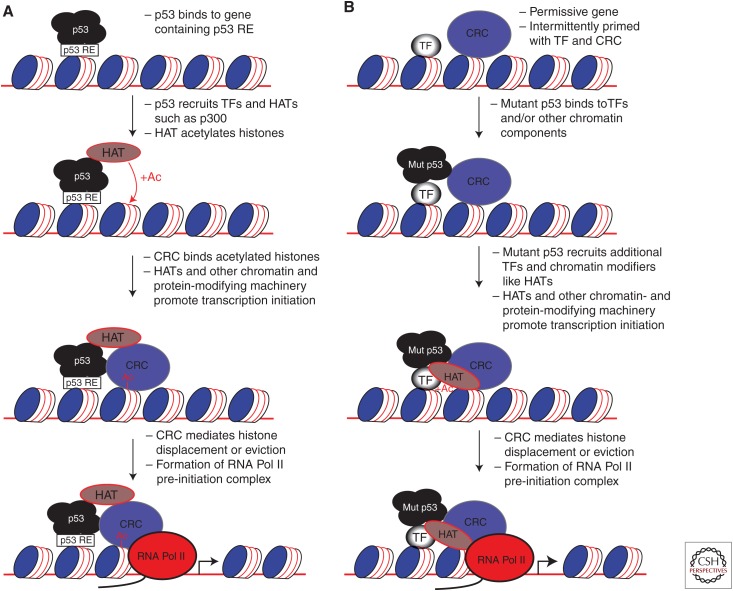
TP53 missense mutations produce a mutant p53 protein that cannot activate the p53 tumor suppressive transcriptional response, which is the primary selective pressure for TP53 mutation. Specific codons of TP53, termed hotspot mutants, are mutated at elevated frequency. Hotspot forms of mutant p53 possess oncogenic properties in addition to being deficient in tumor suppression. Such p53 mutants accumulate to high levels in the cells they inhabit, causing transcriptional alterations that produce pro-oncogenic activities, such as increased pro-growth signaling, invasiveness, and metastases. These forms of mutant p53 very likely use features of wild-type p53, such as interactions with the transcriptional machinery, to produce oncogenic effects. In this review, we discuss commonalities between wild-type and mutant p53 proteins with an emphasis on transcriptional processes.
Copyright © 2017 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. 2009. A mutant-p53/Smad complex opposes p63 to empower TGF-β-induced metastasis. Cell 137: 87–98. - PubMed
-
- Agalioti T, Chen G, Thanos D. 2002. Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381–392. - PubMed
-
- Ahrendt SA, Hu Y, Buta M, McDermott MP, Benoit N, Yang SC, Wu L, Sidransky D. 2003. p53 mutations and survival in stage I non-small-cell lung cancer: Results of a prospective study. J Natl Cancer Inst 95: 961–970. - PubMed
-
- Alsner J, Jensen V, Kyndi M, Offersen BV, Vu P, Borresen-Dale AL, Overgaard J. 2008. A comparison between p53 accumulation determined by immunohistochemistry and TP53 mutations as prognostic variables in tumours from breast cancer patients. Acta Oncol 47: 600–607. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous