

Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho
- PMID: 27837149
- PMCID: PMC5373441
- DOI: 10.1681/ASN.2015111266
Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho
Abstract
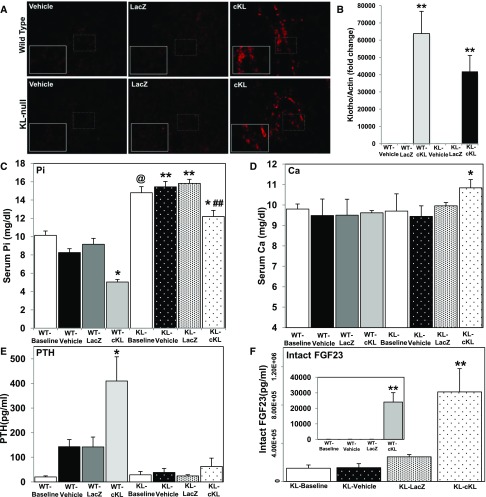
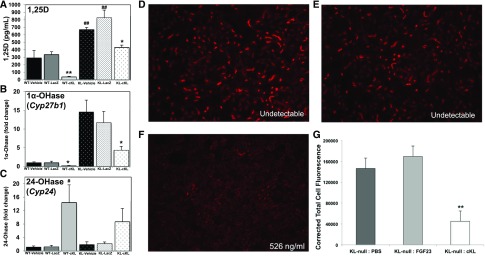
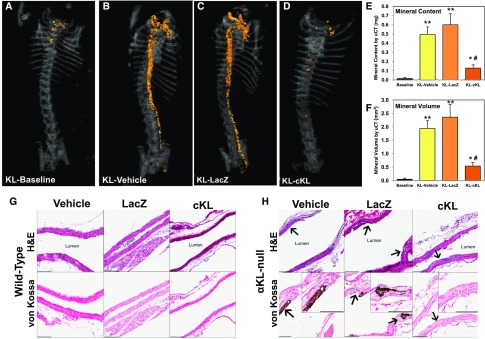
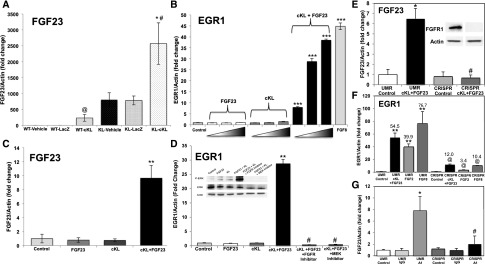
αKlotho (αKL) regulates mineral metabolism, and diseases associated with αKL deficiency are characterized by hyperphosphatemia and vascular calcification (VC). αKL is expressed as a membrane-bound protein (mKL) and recognized as the coreceptor for fibroblast growth factor-23 (FGF23) and a circulating soluble form (cKL) created by endoproteolytic cleavage of mKL. The functions of cKL with regard to phosphate metabolism are unclear. We tested the ability of cKL to regulate pathways and phenotypes associated with hyperphosphatemia in a mouse model of CKD-mineral bone disorder and αKL-null mice. Stable delivery of adeno-associated virus (AAV) expressing cKL to diabetic endothelial nitric oxide synthase-deficient mice or αKL-null mice reduced serum phosphate levels. Acute injection of recombinant cKL downregulated the renal sodium-phosphate cotransporter Npt2a in αKL-null mice supporting direct actions of cKL in the absence of mKL. αKL-null mice with sustained AAV-cKL expression had a 74%-78% reduction in aorta mineral content and a 72%-77% reduction in mineral volume compared with control-treated counterparts (P<0.01). Treatment of UMR-106 osteoblastic cells with cKL + FGF23 increased the phosphorylation of extracellular signal-regulated kinase 1/2 and induced Fgf23 expression. CRISPR/Cas9-mediated deletion of fibroblast growth factor receptor 1 (FGFR1) or pretreatment with inhibitors of mitogen-activated kinase kinase 1 or FGFR ablated these responses. In summary, sustained cKL treatment reduced hyperphosphatemia in a mouse model of CKD-mineral bone disorder, and it reduced hyperphosphatemia and prevented VC in mice without endogenous αKL. Furthermore, cKL stimulated Fgf23 in an FGFR1-dependent manner in bone cells. Collectively, these findings indicate that cKL has mKL-independent activity and suggest the potential for enhancing cKL activity in diseases of hyperphosphatemia with associated VC.
Keywords: FGF23; alpha-klotho; bone; db/db-eNOS; hyperphosphatemia; osteocyte.
Copyright © 2017 by the American Society of Nephrology.
Figures

References
-
- Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F, Kuen E, König P, Kraatz G, Mann JF, Müller GA, Köhler H, Riegler P, Riegler P: Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 18: 2600–2608, 2007 - PubMed
-
- Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE: Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 207: 546–551, 2009 - PubMed
-
- Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, Zhang X, Nessel L, Hamano T, Grunwald JE, Raj DS, Yang W, He J, Lash JP, Go AS, Kusek JW, Feldman H, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Investigators : Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol 25: 349–360, 2014 - PMC - PubMed
-
- Ix JH, Katz R, Kestenbaum BR, de Boer IH, Chonchol M, Mukamal KJ, Rifkin D, Siscovick DS, Sarnak MJ, Shlipak MG: Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol 60: 200–207, 2012 - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
