

Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography
- PMID: 27841871
- PMCID: PMC5502819
- DOI: 10.1038/nature20599
Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography
Abstract
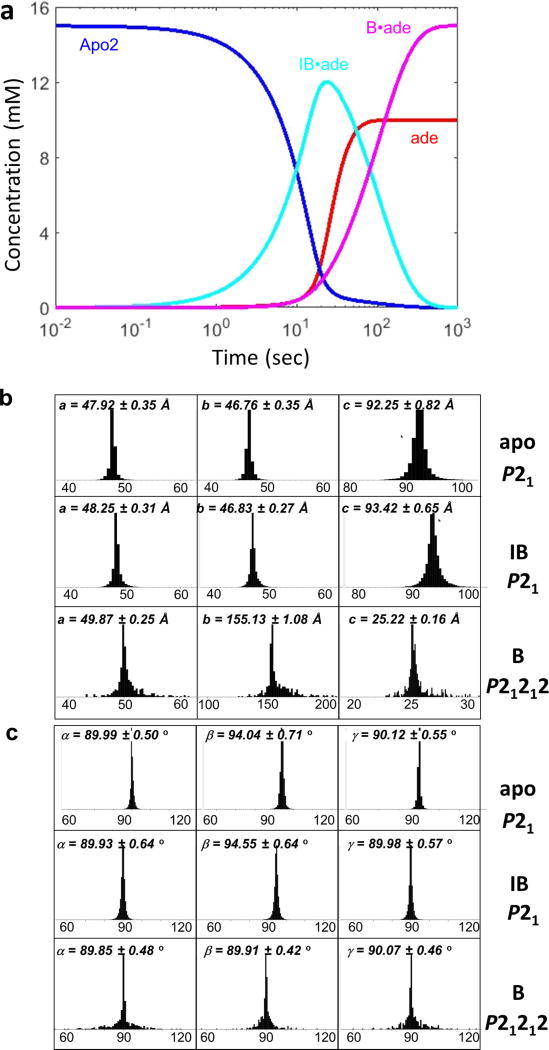
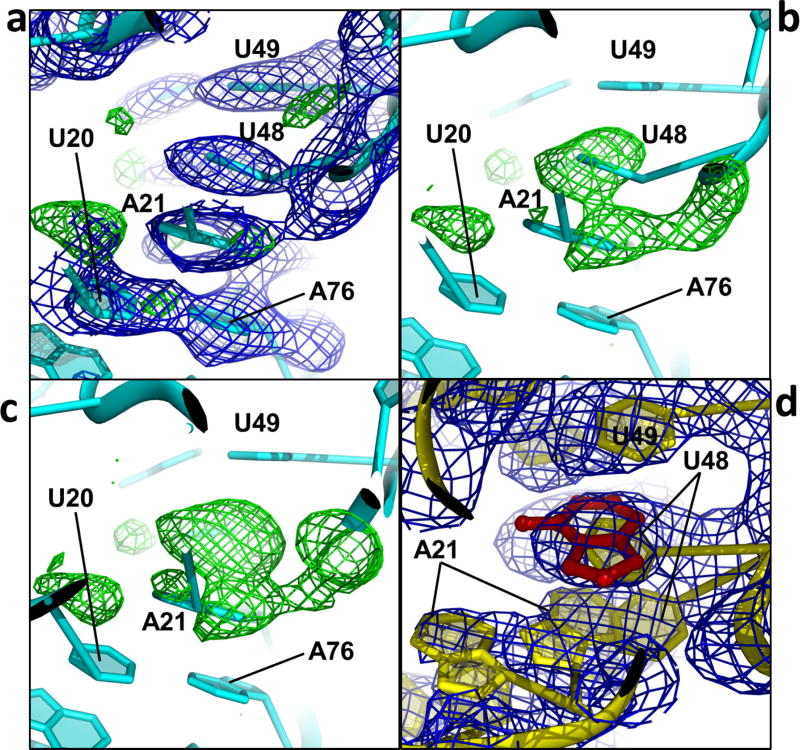
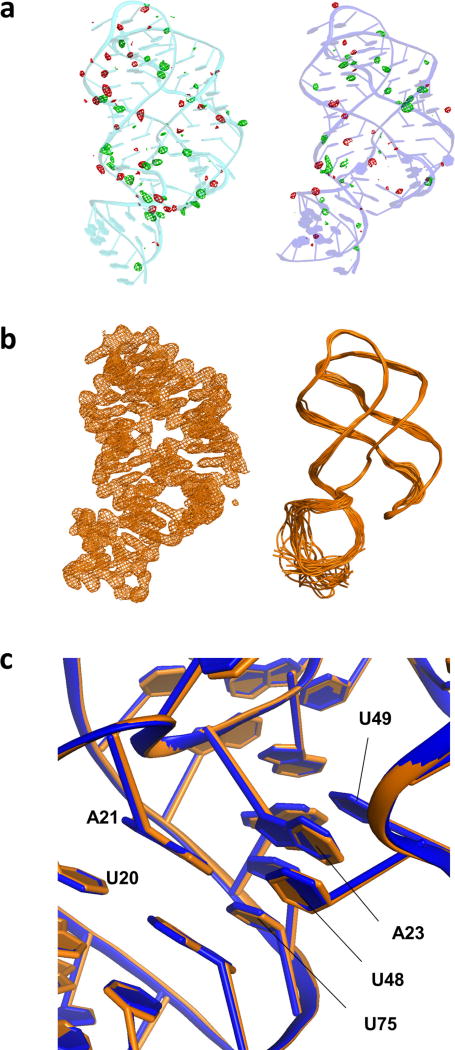
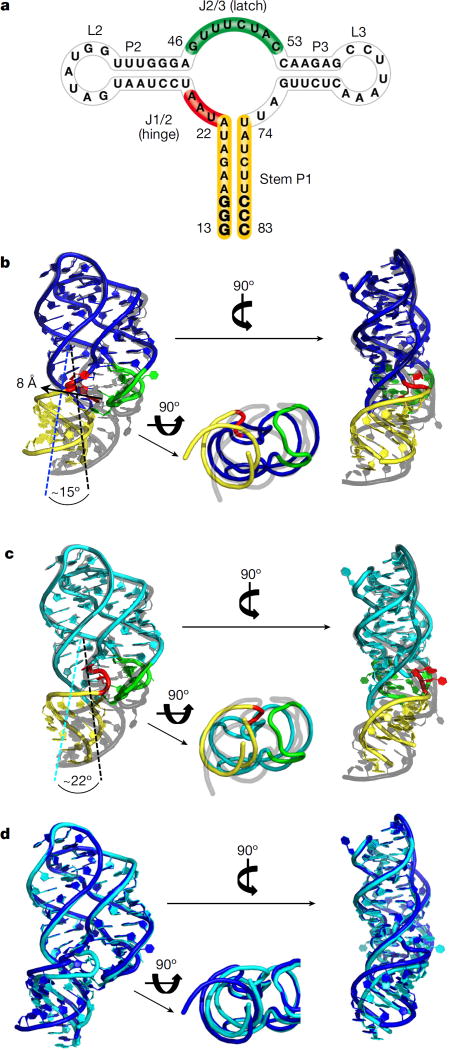
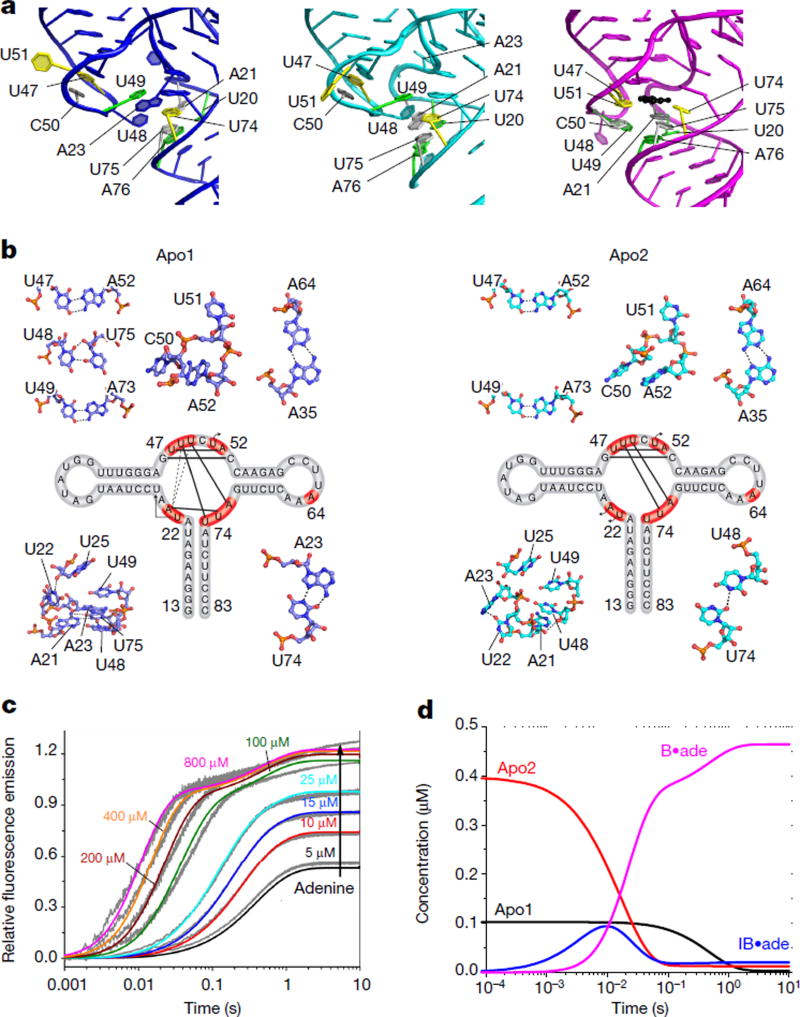
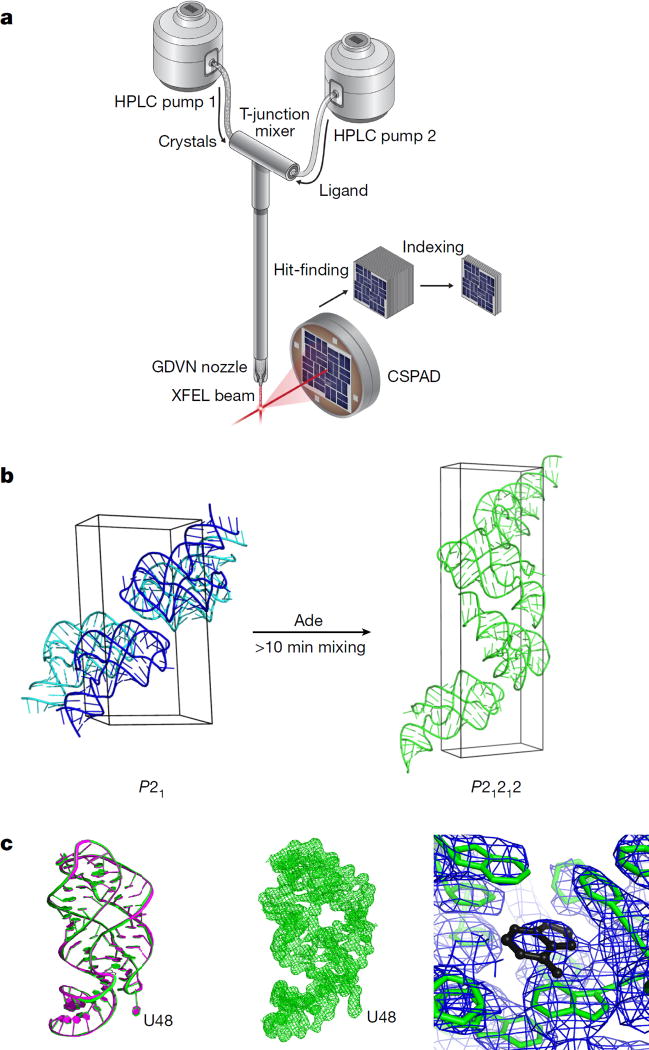
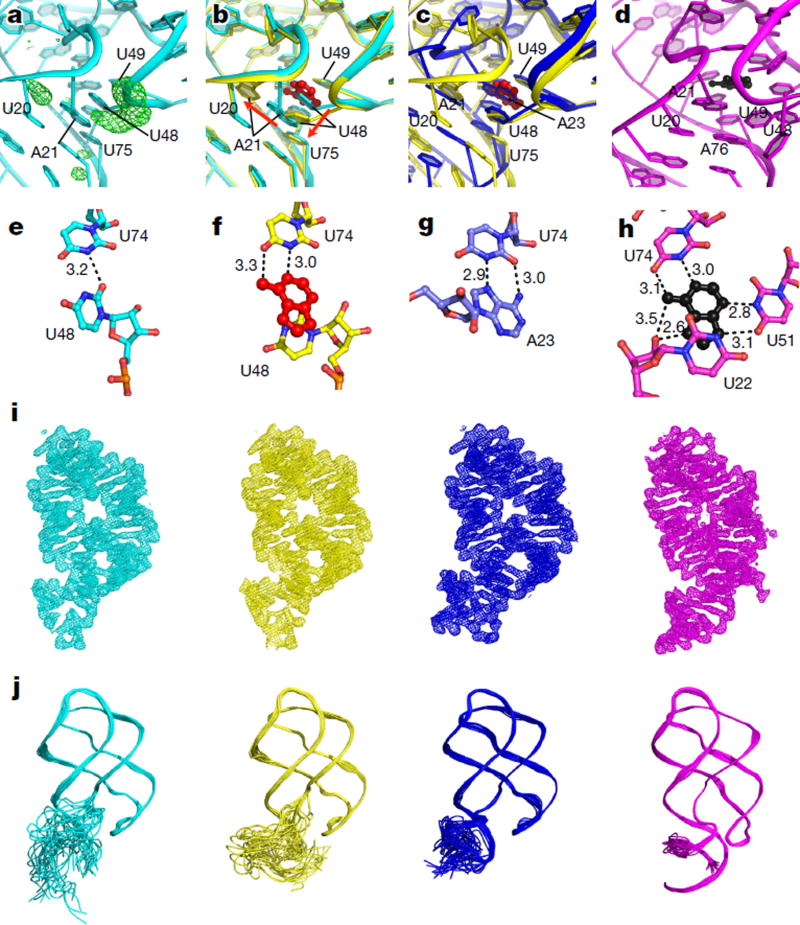
Riboswitches are structural RNA elements that are generally located in the 5' untranslated region of messenger RNA. During regulation of gene expression, ligand binding to the aptamer domain of a riboswitch triggers a signal to the downstream expression platform. A complete understanding of the structural basis of this mechanism requires the ability to study structural changes over time. Here we use femtosecond X-ray free electron laser (XFEL) pulses to obtain structural measurements from crystals so small that diffusion of a ligand can be timed to initiate a reaction before diffraction. We demonstrate this approach by determining four structures of the adenine riboswitch aptamer domain during the course of a reaction, involving two unbound apo structures, one ligand-bound intermediate, and the final ligand-bound conformation. These structures support a reaction mechanism model with at least four states and illustrate the structural basis of signal transmission. The three-way junction and the P1 switch helix of the two apo conformers are notably different from those in the ligand-bound conformation. Our time-resolved crystallographic measurements with a 10-second delay captured the structure of an intermediate with changes in the binding pocket that accommodate the ligand. With at least a 10-minute delay, the RNA molecules were fully converted to the ligand-bound state, in which the substantial conformational changes resulted in conversion of the space group. Such notable changes in crystallo highlight the important opportunities that micro- and nanocrystals may offer in these and similar time-resolved diffraction studies. Together, these results demonstrate the potential of 'mix-and-inject' time-resolved serial crystallography to study biochemically important interactions between biomacromolecules and ligands, including those that involve large conformational changes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
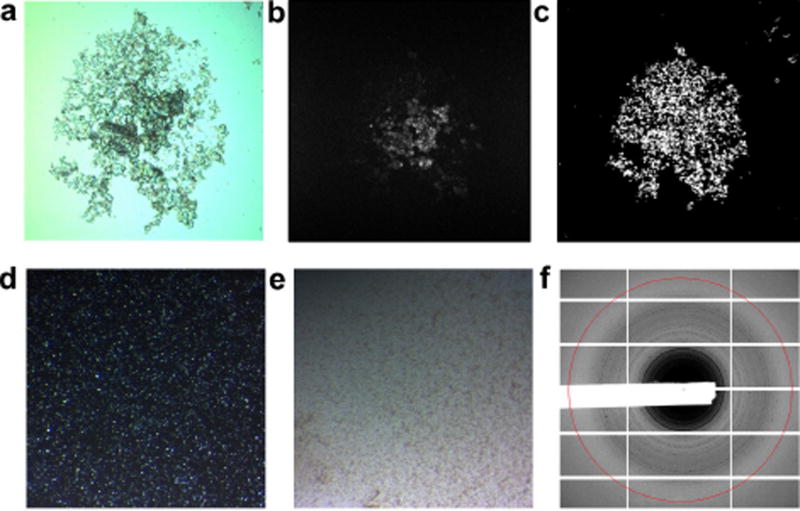
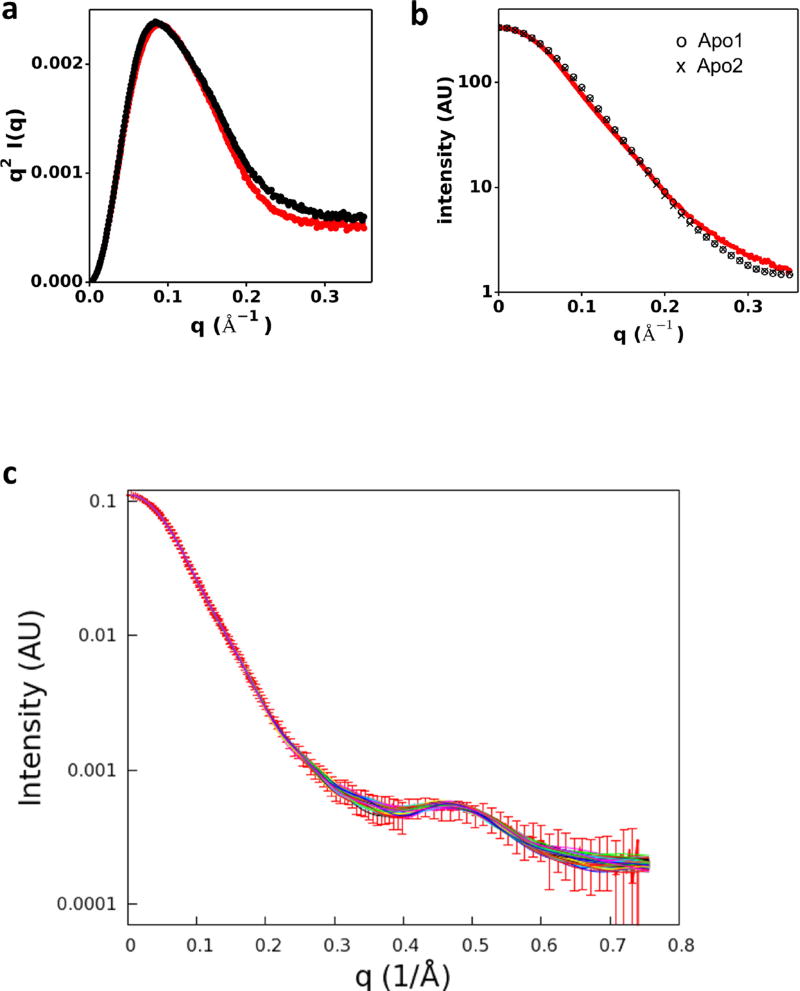
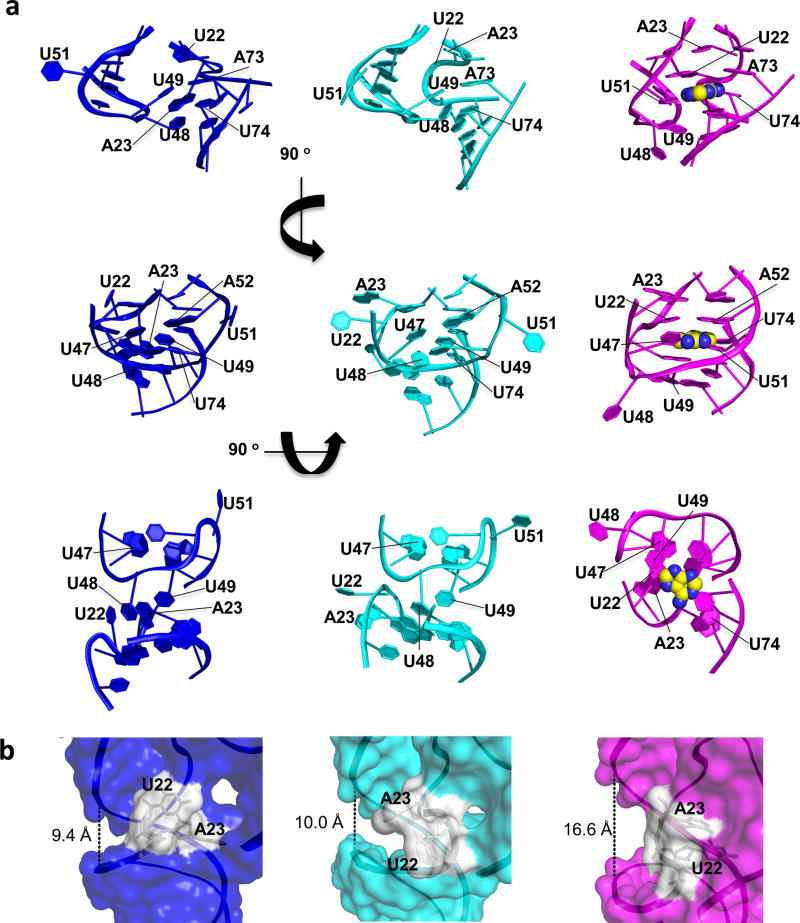
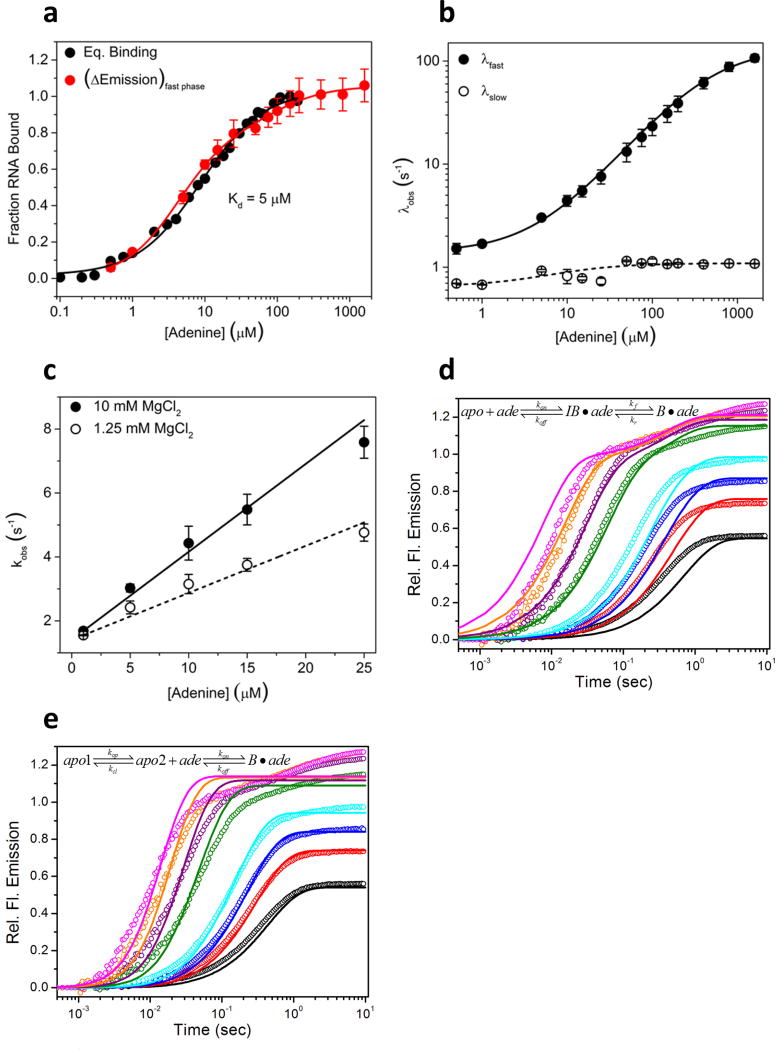
References
-
- Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003;113:577–586. - PubMed
-
- Nahvi A, et al. Genetic control by a metabolite binding mRNA. Chem. Biol. 2002;9:1043. - PubMed
-
- Hajdu J, et al. Analyzing protein functions in four dimensions. Nat. Struct. Biol. 2000;7:1006–1012. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
