

Nitroxidative Signaling Mechanisms in Pathological Pain
- PMID: 27842920
- PMCID: PMC5148691
- DOI: 10.1016/j.tins.2016.10.003
Nitroxidative Signaling Mechanisms in Pathological Pain
Abstract
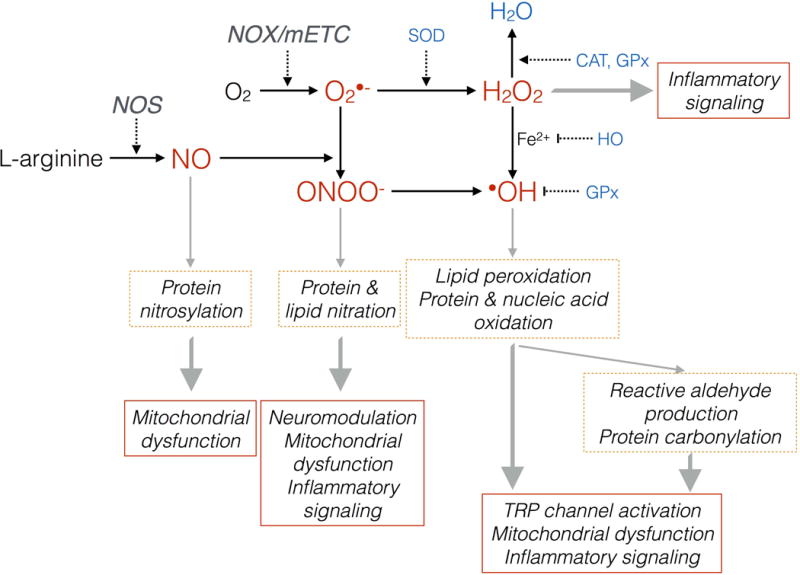
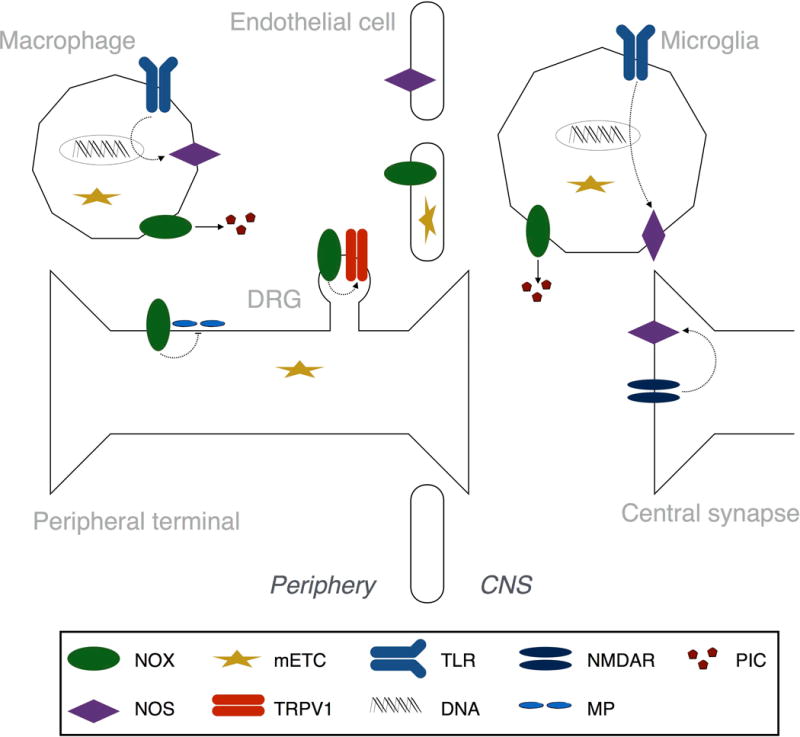
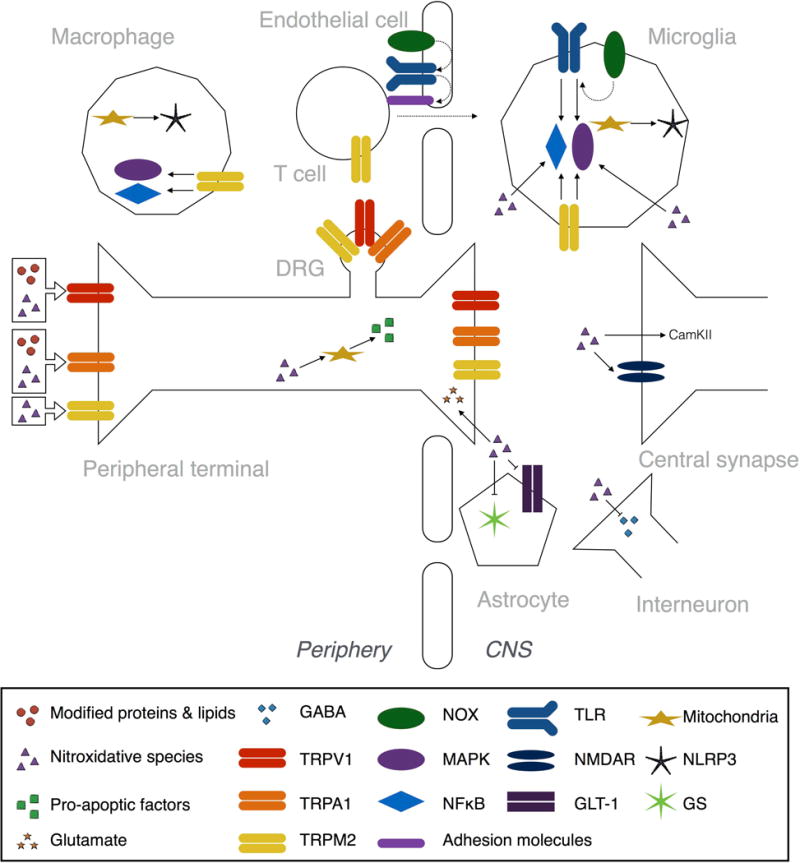
Tissue injury can initiate bidirectional signaling between neurons, glia, and immune cells that creates and amplifies pain. While the ability for neurotransmitters, neuropeptides, and cytokines to initiate and maintain pain has been extensively studied, recent work has identified a key role for reactive oxygen and nitrogen species (ROS/RNS; nitroxidative species), including superoxide, peroxynitrite, and hydrogen peroxide. In this review we describe how nitroxidative species are generated after tissue injury and the mechanisms by which they enhance neuroexcitability in pain pathways. Finally, we discuss potential therapeutic strategies for normalizing nitroxidative signaling, which may also enhance opioid analgesia, to help to alleviate the enormous burden of pathological pain.
Keywords: NADPH oxidase; TRP channels; exercise; mitochondria; neuroinflammation; sensitization.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain.Amino Acids. 2012 Jan;42(1):75-94. doi: 10.1007/s00726-010-0633-0. Epub 2010 Jun 16. Amino Acids. 2012. PMID: 20552384 Review.
-
Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function.Br J Pharmacol. 2017 Jun;174(12):1670-1689. doi: 10.1111/bph.13403. Epub 2016 Feb 4. Br J Pharmacol. 2017. PMID: 26660451 Free PMC article. Review.
-
Signaling of reactive oxygen and nitrogen species in Diabetes mellitus.Oxid Med Cell Longev. 2010 Nov-Dec;3(6):361-73. doi: 10.4161/oxim.3.6.14415. Epub 2010 Nov 1. Oxid Med Cell Longev. 2010. PMID: 21311214 Free PMC article.
-
Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle.Antioxid Redox Signal. 2014 Feb 20;20(6):929-36. doi: 10.1089/ars.2013.5517. Epub 2014 Jan 3. Antioxid Redox Signal. 2014. PMID: 23971496 Free PMC article. Review.
-
Roles of reactive oxygen and nitrogen species in pain.Free Radic Biol Med. 2011 Sep 1;51(5):951-66. doi: 10.1016/j.freeradbiomed.2011.01.026. Epub 2011 Jan 28. Free Radic Biol Med. 2011. PMID: 21277369 Free PMC article. Review.
Cited by
-
Pain in Hemophilia: Unexplored Role of Oxidative Stress.Antioxidants (Basel). 2022 Jun 3;11(6):1113. doi: 10.3390/antiox11061113. Antioxidants (Basel). 2022. PMID: 35740010 Free PMC article. Review.
-
Acute Effects of Brief Mindfulness Intervention Coupled with Carbohydrate Ingestion to Re-Energize Soccer Players: A Randomized Crossover Trial.Int J Environ Res Public Health. 2020 Dec 4;17(23):9037. doi: 10.3390/ijerph17239037. Int J Environ Res Public Health. 2020. PMID: 33291535 Free PMC article. Clinical Trial.
-
Spinal Reactive Oxygen Species and Oxidative Damage Mediate Chronic Pain in Lame Dairy Cows.Animals (Basel). 2019 Sep 17;9(9):693. doi: 10.3390/ani9090693. Animals (Basel). 2019. PMID: 31533257 Free PMC article.
-
A role for neuroimmune signaling in a rat model of Gulf War Illness-related pain.Brain Behav Immun. 2021 Jan;91:418-428. doi: 10.1016/j.bbi.2020.10.022. Epub 2020 Oct 27. Brain Behav Immun. 2021. PMID: 33127584 Free PMC article.
-
Synthetic Secoisolariciresinol Diglucoside Attenuates Established Pain, Oxidative Stress and Neuroinflammation in a Rodent Model of Painful Radiculopathy.Antioxidants (Basel). 2020 Nov 30;9(12):1209. doi: 10.3390/antiox9121209. Antioxidants (Basel). 2020. PMID: 33266301 Free PMC article.
References
-
- Krebs HA, Johnson WA. The role of citric acid in intermediate metabolism in animal tissues. Enzymologia. 1937;4:148–156. - PubMed
-
- Gozsy B, Szent-Gyorgyi A. On the mechanism of primary respiration in pigeon breast muscle. Hoppe Seylers Z Physiol Chem. 1934;224:1–10.
-
- Harden A, Young WJ. The Alcoholic Ferment of Yeast-Juice. Proc R Soc Lond B Biol Sci. 1906;77:405–420.
-
- Maass O, Hatcher WH. The properties of pure hydrogen peroxide. I. J Am Chem Soc. 1920;42:2548–69.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
