

Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage
- PMID: 27846393
- PMCID: PMC5805090
- DOI: 10.1016/j.ccell.2016.09.014
Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage
Abstract
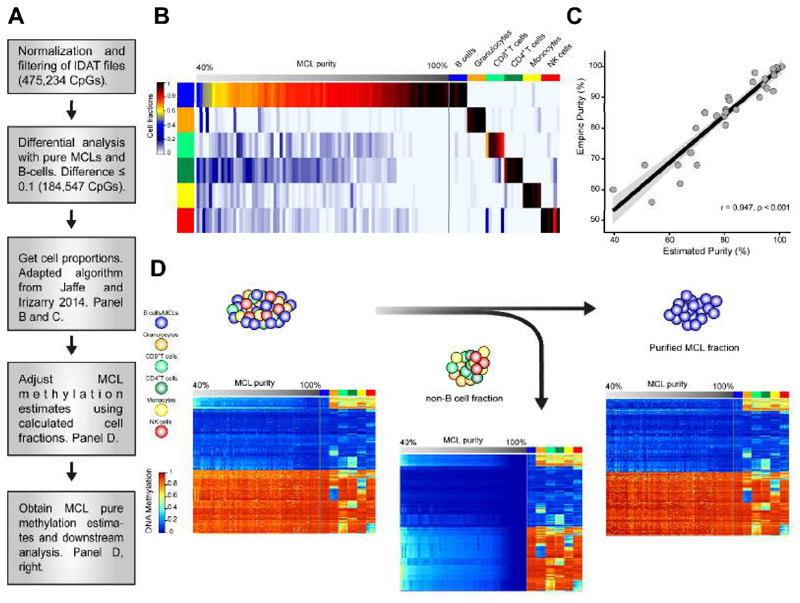
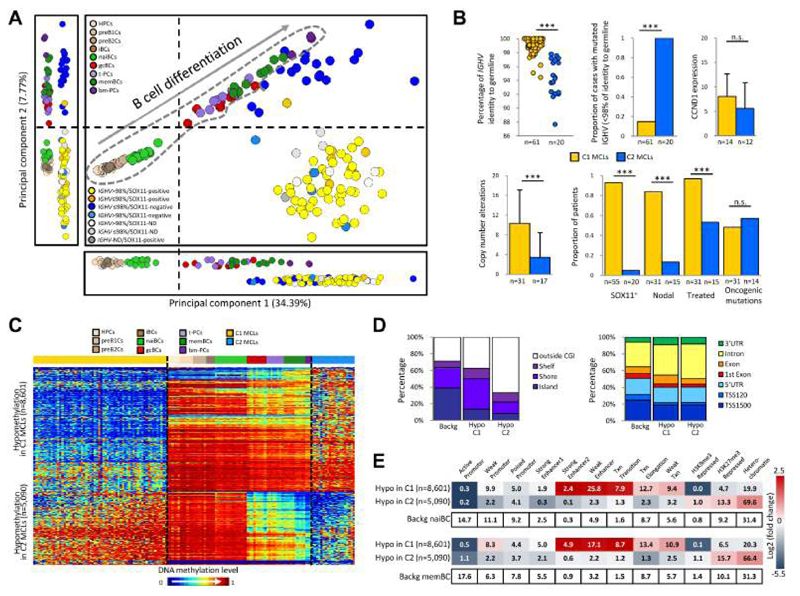
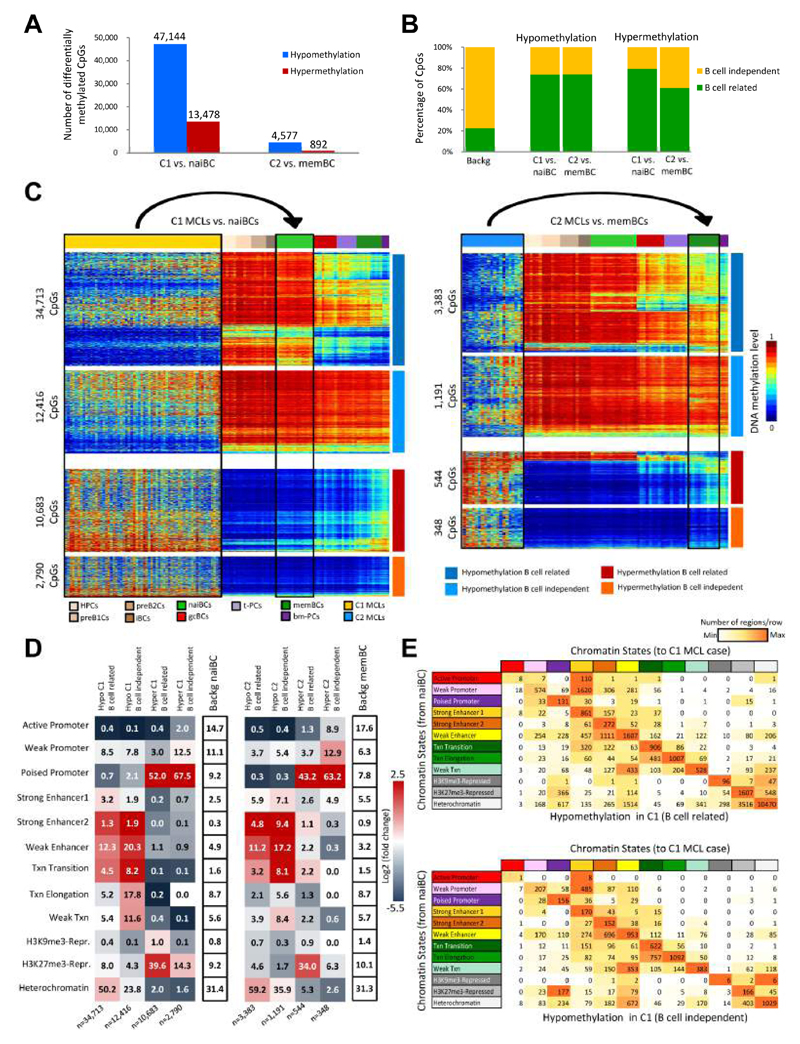
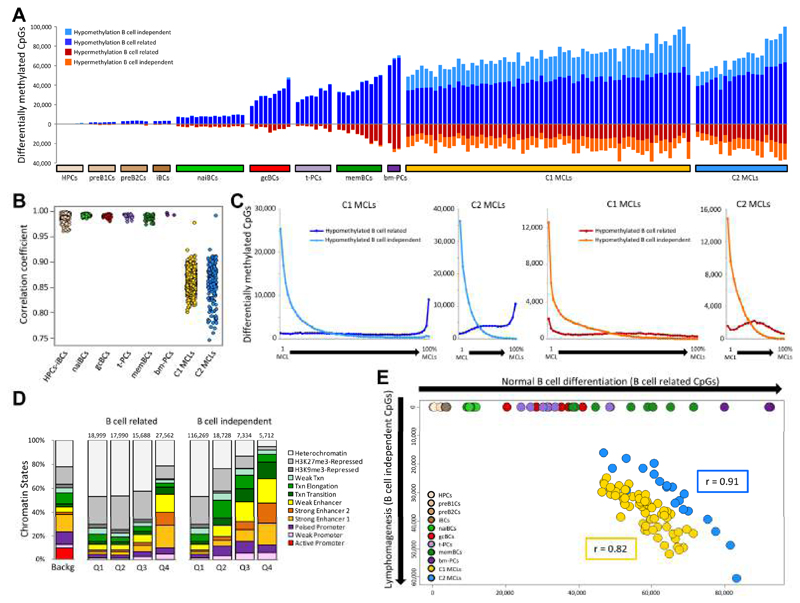
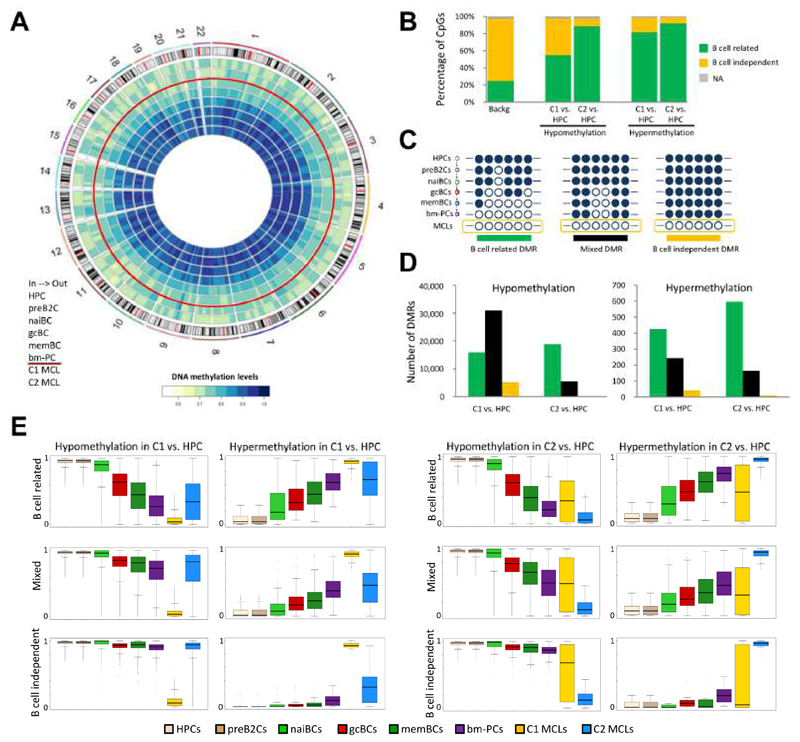
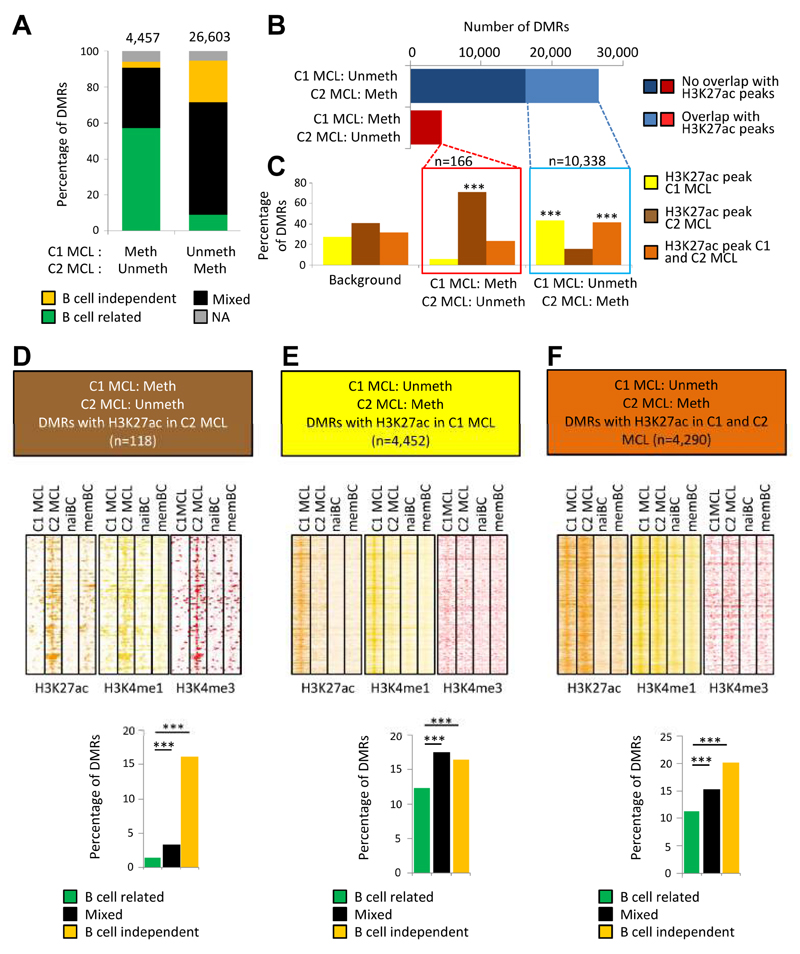
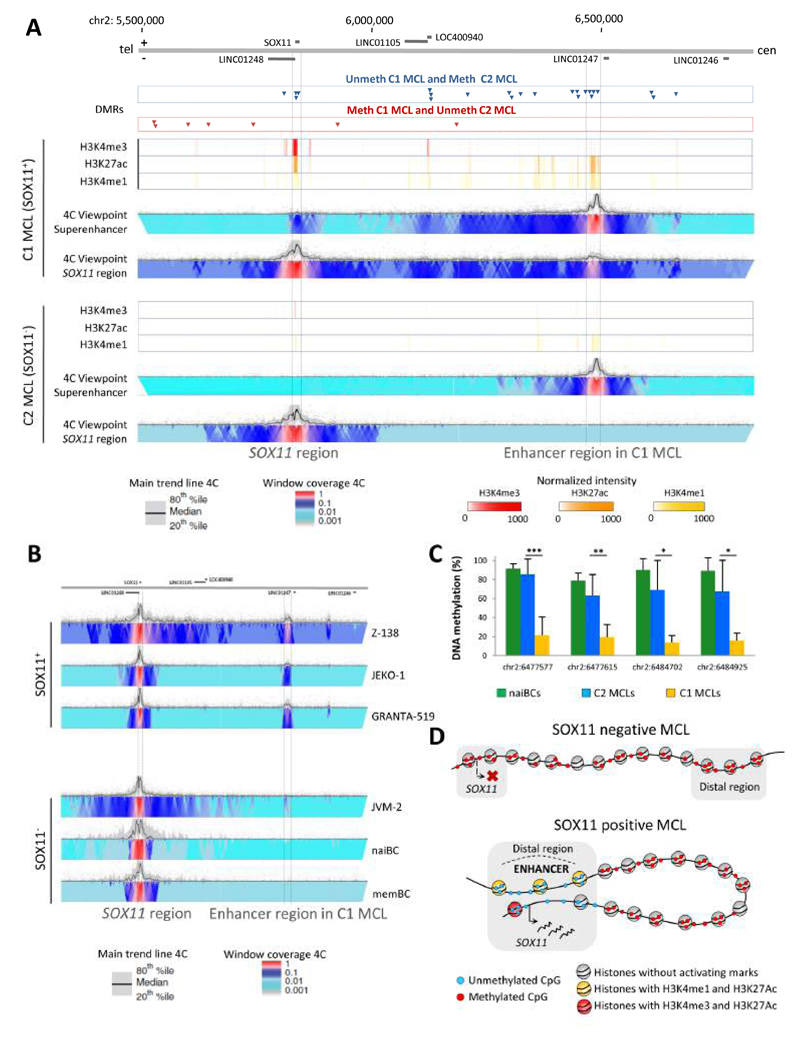
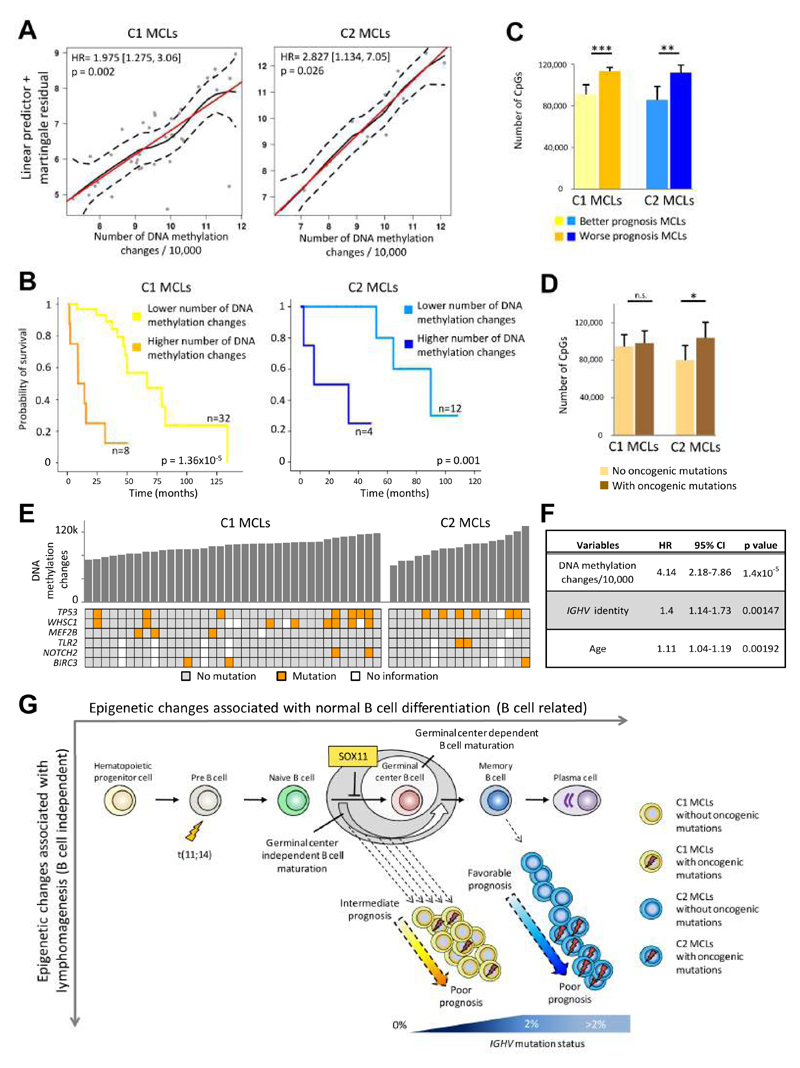
We analyzed the in silico purified DNA methylation signatures of 82 mantle cell lymphomas (MCL) in comparison with cell subpopulations spanning the entire B cell lineage. We identified two MCL subgroups, respectively carrying epigenetic imprints of germinal-center-inexperienced and germinal-center-experienced B cells, and we found that DNA methylation profiles during lymphomagenesis are largely influenced by the methylation dynamics in normal B cells. An integrative epigenomic approach revealed 10,504 differentially methylated regions in regulatory elements marked by H3K27ac in MCL primary cases, including a distant enhancer showing de novo looping to the MCL oncogene SOX11. Finally, we observed that the magnitude of DNA methylation changes per case is highly variable and serves as an independent prognostic factor for MCL outcome.
Keywords: ChIP-seq; DNA looping; DNA methylation; SOX11; chromatin; enhancer; epigenomics; lymphoma; mantle cell lymphoma; whole-genome bisulfite sequencing.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Comment on
-
Unraveling the DNA Methylome in Mantle Cell Lymphoma: New Insights into the Cellular Origin.Cancer Cell. 2016 Nov 14;30(5):665-667. doi: 10.1016/j.ccell.2016.10.013. Cancer Cell. 2016. PMID: 27846388
References
-
- Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, Bock C, Boehm B, Campo E, Caricasole A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30:224–226. - PubMed
-
- Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
