

Guidance for the utility of linear models in meta-analysis of genetic association studies of binary phenotypes
- PMID: 27848946
- PMCID: PMC5237383
- DOI: 10.1038/ejhg.2016.150
Guidance for the utility of linear models in meta-analysis of genetic association studies of binary phenotypes
Abstract
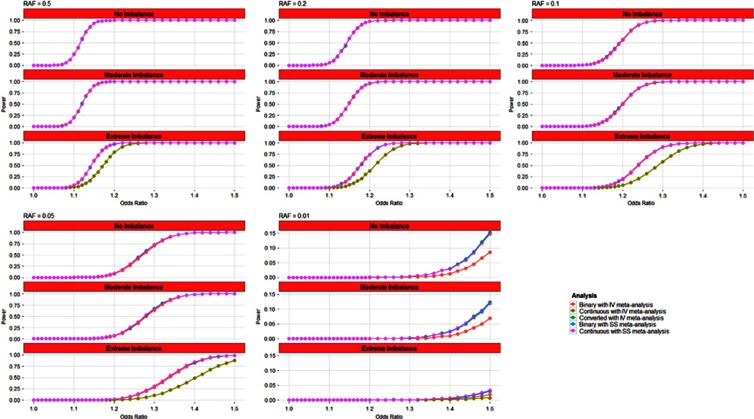
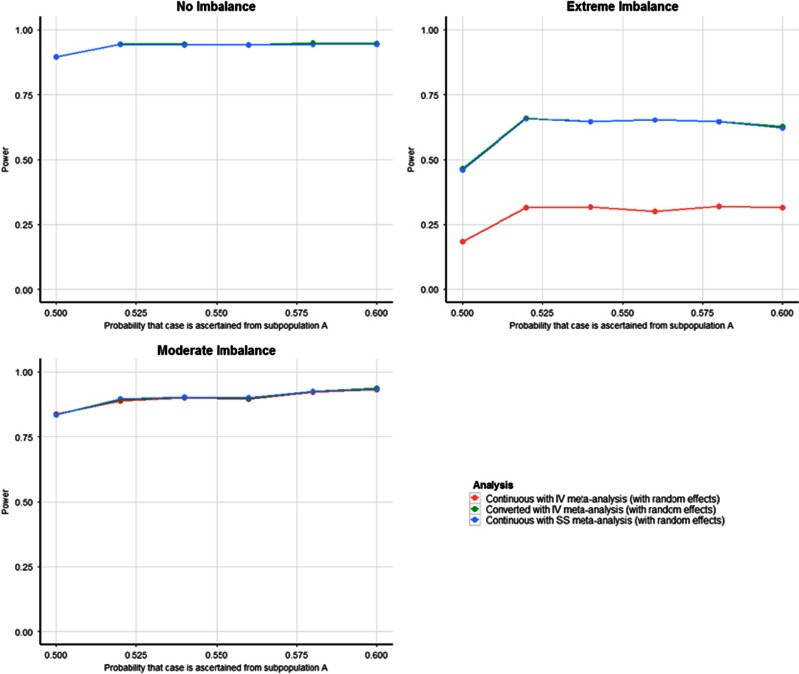
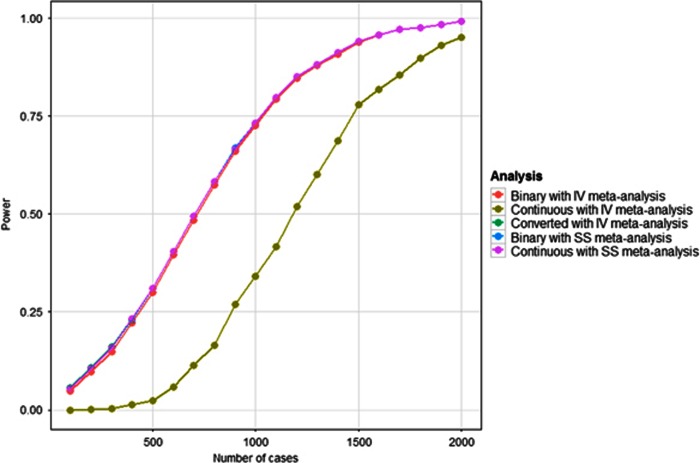
Linear mixed models are increasingly used for the analysis of genome-wide association studies (GWAS) of binary phenotypes because they can efficiently and robustly account for population stratification and relatedness through inclusion of random effects for a genetic relationship matrix. However, the utility of linear (mixed) models in the context of meta-analysis of GWAS of binary phenotypes has not been previously explored. In this investigation, we present simulations to compare the performance of linear and logistic regression models under alternative weighting schemes in a fixed-effects meta-analysis framework, considering designs that incorporate variable case-control imbalance, confounding factors and population stratification. Our results demonstrate that linear models can be used for meta-analysis of GWAS of binary phenotypes, without loss of power, even in the presence of extreme case-control imbalance, provided that one of the following schemes is used: (i) effective sample size weighting of Z-scores or (ii) inverse-variance weighting of allelic effect sizes after conversion onto the log-odds scale. Our conclusions thus provide essential recommendations for the development of robust protocols for meta-analysis of binary phenotypes with linear models.
Conflict of interest statement
Conflict of interest. The authors declare no conflict of interest.
Figures
References
-
- Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D: FaST linear mixed models for genome-wide association studies. Nat Methods 2011; 8: 833–835. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
