

Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling
- PMID: 27852822
- PMCID: PMC5207144
- DOI: 10.1074/jbc.M116.754887
Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling
Abstract
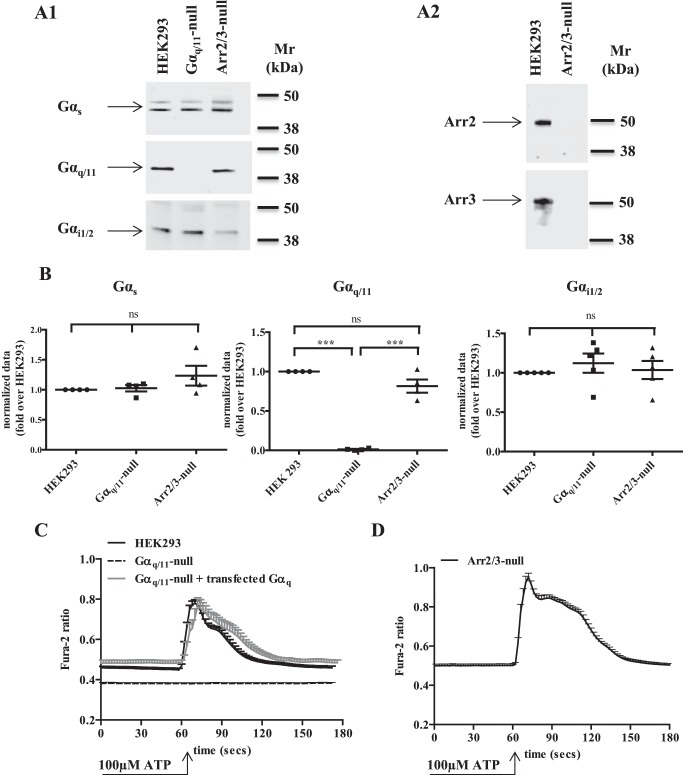
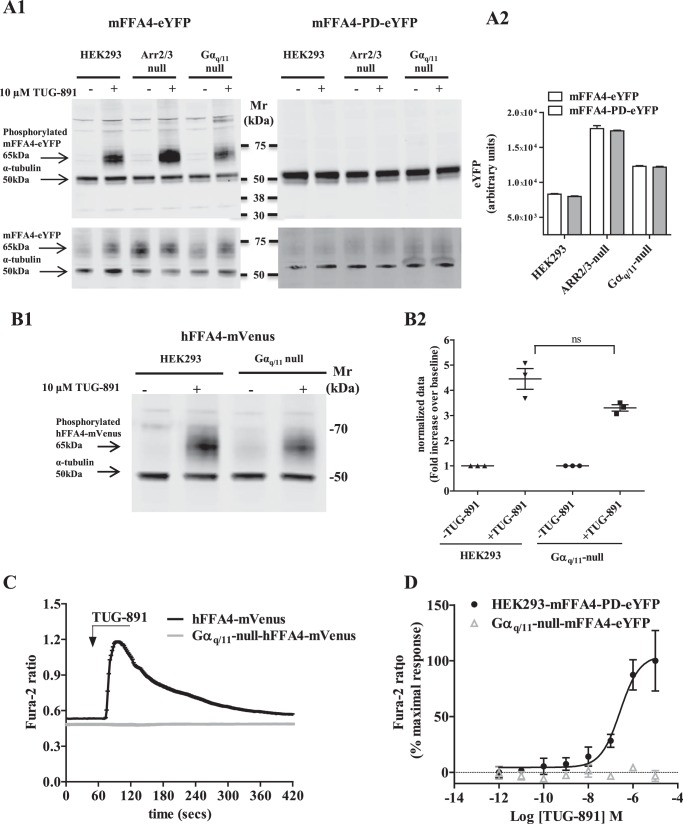
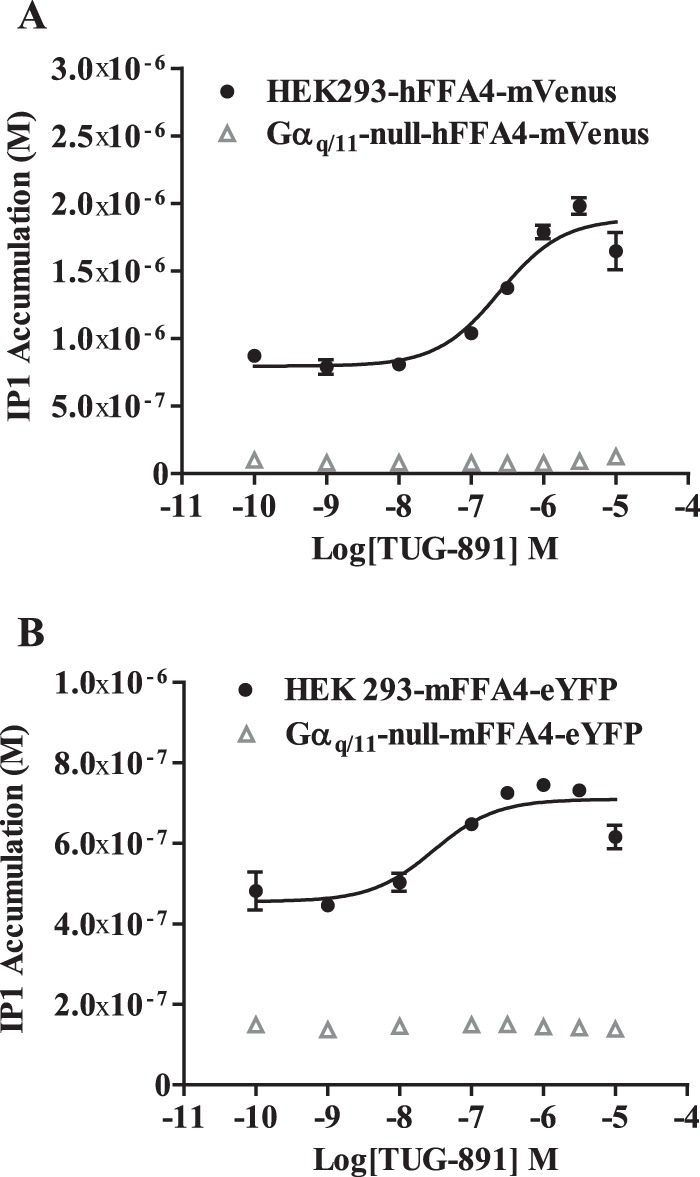
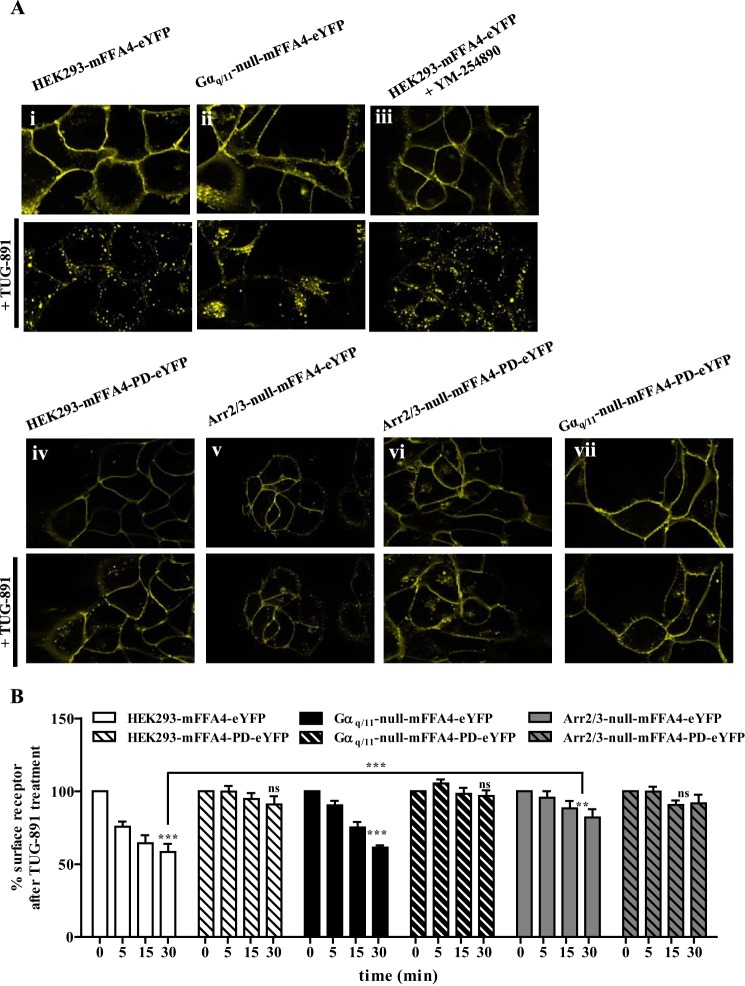
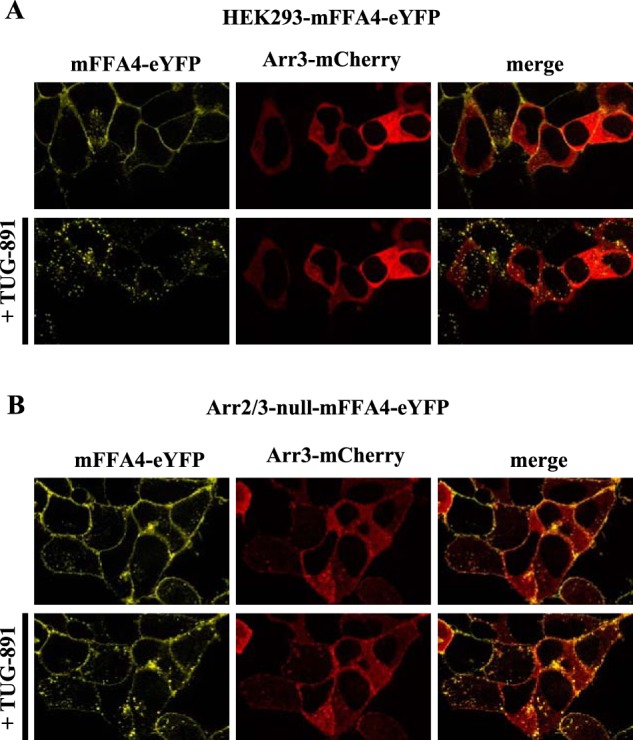
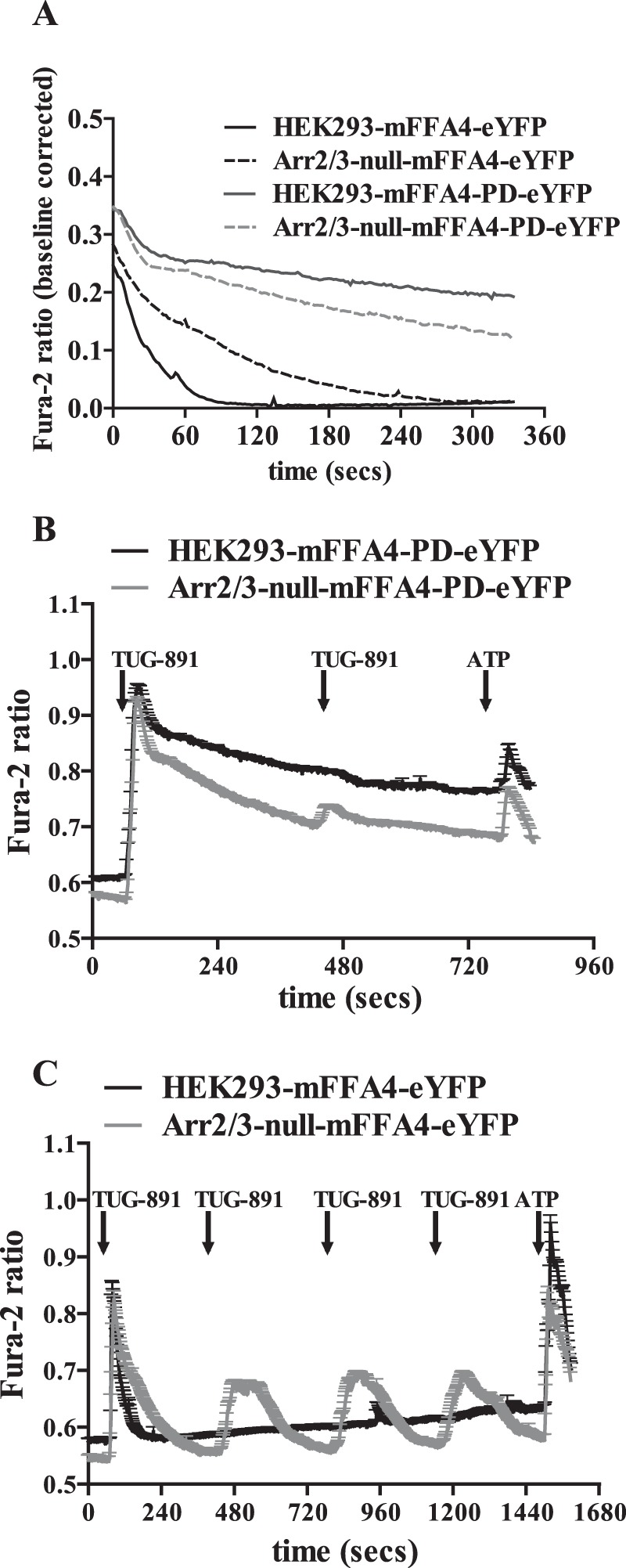
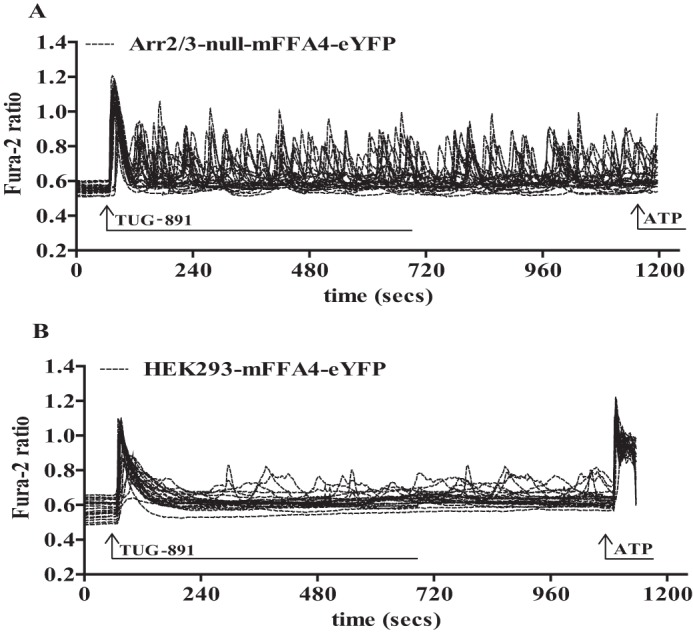
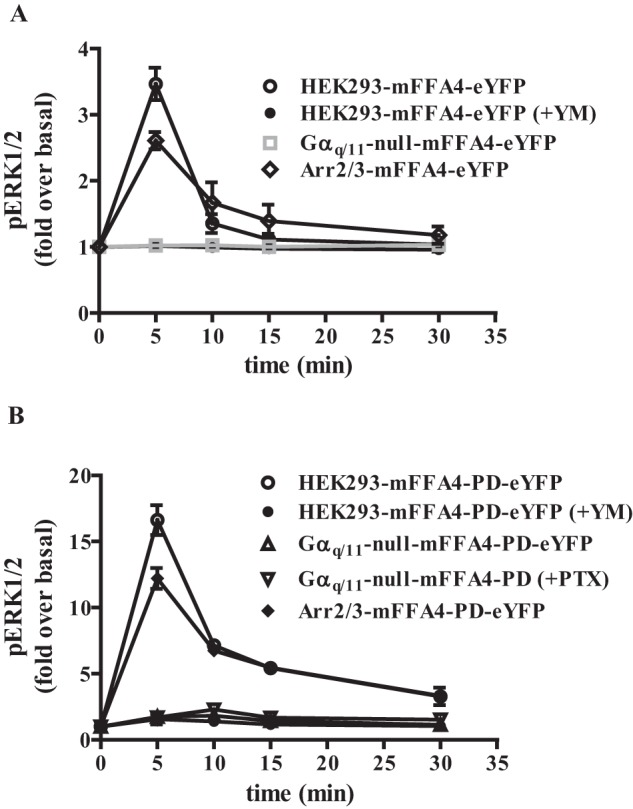
G protein-coupled receptors (GPCRs) can initiate intracellular signaling cascades by coupling to an array of heterotrimeric G proteins and arrestin adaptor proteins. Understanding the contribution of each of these coupling options to GPCR signaling has been hampered by a paucity of tools to selectively perturb receptor function. Here we employ CRISPR/Cas9 genome editing to eliminate selected G proteins (Gαq and Gα11) or arrestin2 and arrestin3 from HEK293 cells together with the elimination of receptor phosphorylation sites to define the relative contribution of G proteins, arrestins, and receptor phosphorylation to the signaling outcomes of the free fatty acid receptor 4 (FFA4). A lack of FFA4-mediated elevation of intracellular Ca2+ in Gαq/Gα11-null cells and agonist-mediated receptor internalization in arrestin2/3-null cells confirmed previously reported canonical signaling features of this receptor, thereby validating the genome-edited HEK293 cells. FFA4-mediated ERK1/2 activation was totally dependent on Gq/11 but intriguingly was substantially enhanced for FFA4 receptors lacking sites of regulated phosphorylation. This was not due to a simple lack of desensitization of Gq/11 signaling because the Gq/11-dependent calcium response was desensitized by both receptor phosphorylation and arrestin-dependent mechanisms, whereas a substantially enhanced ERK1/2 response was only observed for receptors lacking phosphorylation sites and not in arrestin2/3-null cells. In conclusion, we validate CRISPR/Cas9 engineered HEK293 cells lacking Gq/11 or arrestin2/3 as systems for GPCR signaling research and employ these cells to reveal a previously unappreciated interplay of signaling pathways where receptor phosphorylation can impact on ERK1/2 signaling through a mechanism that is likely independent of arrestins.
Keywords: CRISPR/Cas; G protein; G protein-coupled receptor (GPCR); arrestin; calcium intracellular release; extracellular-signal-regulated kinase (ERK); fatty acid.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Lefkowitz RJ. (2013) A brief history of G-protein-coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 52, 6366–6378 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
