

Animal models of β-hemoglobinopathies: utility and limitations
- PMID: 27853395
- PMCID: PMC5104300
- DOI: 10.2147/JBM.S87955
Animal models of β-hemoglobinopathies: utility and limitations
Abstract
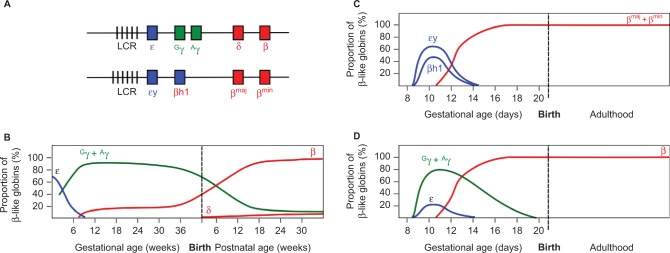
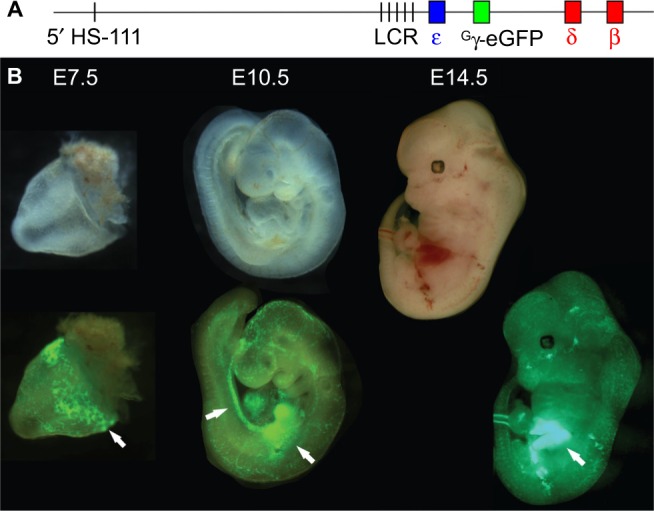
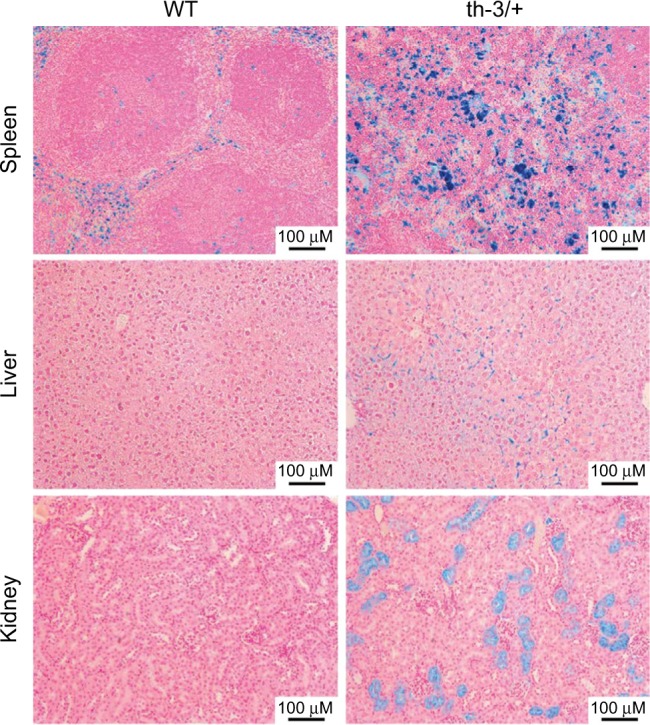
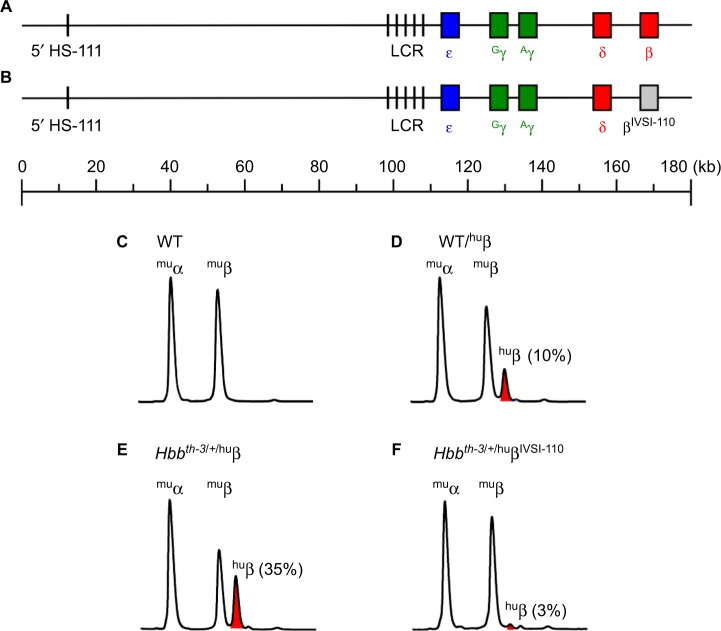
The structural and functional conservation of hemoglobin throughout mammals has made the laboratory mouse an exceptionally useful organism in which to study both the protein and the individual globin genes. Early researchers looked to the globin genes as an excellent model in which to examine gene regulation - bountifully expressed and displaying a remarkably consistent pattern of developmental activation and silencing. In parallel with the growth of research into expression of the globin genes, mutations within the β-globin gene were identified as the cause of the β-hemoglobinopathies such as sickle cell disease and β-thalassemia. These lines of enquiry stimulated the development of transgenic mouse models, first carrying individual human globin genes and then substantial human genomic fragments incorporating the multigenic human β-globin locus and regulatory elements. Finally, mice were devised carrying mutant human β-globin loci on genetic backgrounds deficient in the native mouse globins, resulting in phenotypes of sickle cell disease or β-thalassemia. These years of work have generated a group of model animals that display many features of the β-hemoglobinopathies and provided enormous insight into the mechanisms of gene regulation. Substantive differences in the expression of human and mouse globins during development have also come to light, revealing the limitations of the mouse model, but also providing opportunities to further explore the mechanisms of globin gene regulation. In addition, animal models of β-hemoglobinopathies have demonstrated the feasibility of gene therapy for these conditions, now showing success in human clinical trials. Such models remain in use to dissect the molecular events of globin gene regulation and to identify novel treatments based upon the reactivation of developmentally silenced γ-globin. Here, we describe the development of animal models to investigate globin switching and the β-hemoglobinopathies, a field that has paralleled the emergence of modern molecular biology and clinical genetics.
Keywords: bacterial artificial chromosome; globin switching; green fluorescent protein; locus control region; sickle cell disease; β-Hemoglobinopathies.
Conflict of interest statement
The authors report no conflicts of interest in this work.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
