

Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures
- PMID: 27855702
- PMCID: PMC5114739
- DOI: 10.1186/s13059-016-1092-z
Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures
Erratum in
-
Erratum to: Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures.Genome Biol. 2017 Mar 1;18(1):46. doi: 10.1186/s13059-017-1180-8. Genome Biol. 2017. PMID: 28249590 Free PMC article. No abstract available.
Abstract
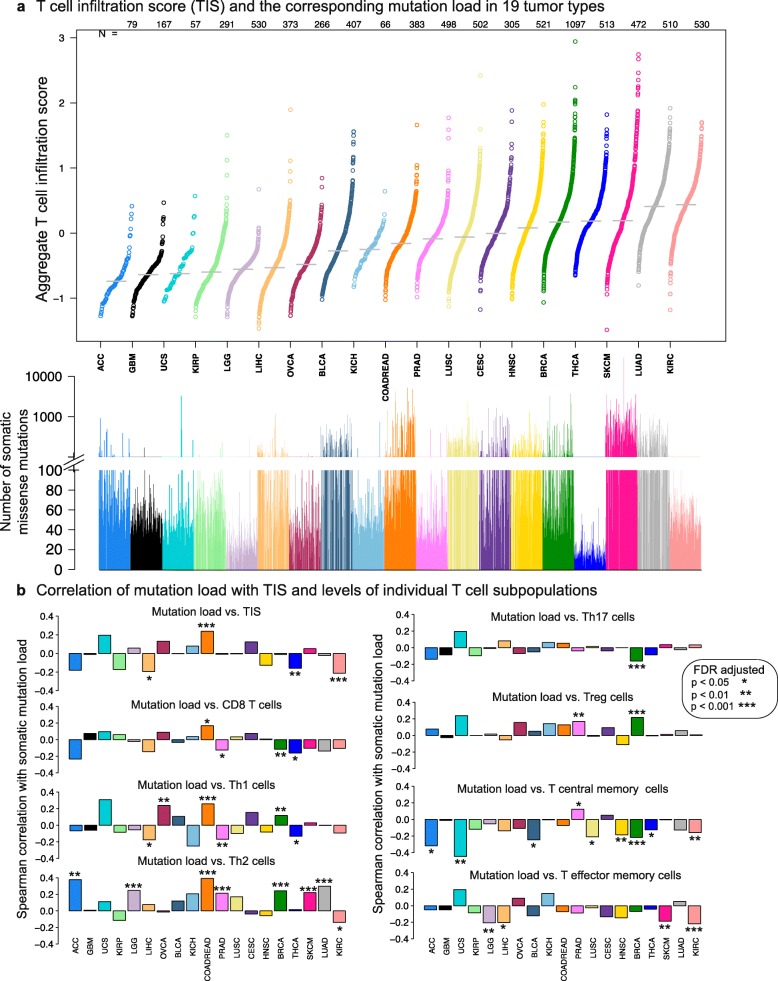
Background: Tumor-infiltrating immune cells have been linked to prognosis and response to immunotherapy; however, the levels of distinct immune cell subsets and the signals that draw them into a tumor, such as the expression of antigen presenting machinery genes, remain poorly characterized. Here, we employ a gene expression-based computational method to profile the infiltration levels of 24 immune cell populations in 19 cancer types.
Results: We compare cancer types using an immune infiltration score and a T cell infiltration score and find that clear cell renal cell carcinoma (ccRCC) is among the highest for both scores. Using immune infiltration profiles as well as transcriptomic and proteomic datasets, we characterize three groups of ccRCC tumors: T cell enriched, heterogeneously infiltrated, and non-infiltrated. We observe that the immunogenicity of ccRCC tumors cannot be explained by mutation load or neo-antigen load, but is highly correlated with MHC class I antigen presenting machinery expression (APM). We explore the prognostic value of distinct T cell subsets and show in two cohorts that Th17 cells and CD8+ T/Treg ratio are associated with improved survival, whereas Th2 cells and Tregs are associated with negative outcomes. Investigation of the association of immune infiltration patterns with the subclonal architecture of tumors shows that both APM and T cell levels are negatively associated with subclone number.
Conclusions: Our analysis sheds light on the immune infiltration patterns of 19 human cancers and unravels mRNA signatures with prognostic utility and immunotherapeutic biomarker potential in ccRCC.
Keywords: Cancer immunotherapy; Checkpoint blockade; Clear cell renal cell carcinoma (ccRCC); Computational deconvolution; Tumor immune microenvironment.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
