

MLL-AF4 binds directly to a BCL-2 specific enhancer and modulates H3K27 acetylation
- PMID: 27856324
- PMCID: PMC5333536
- DOI: 10.1016/j.exphem.2016.11.003
MLL-AF4 binds directly to a BCL-2 specific enhancer and modulates H3K27 acetylation
Abstract
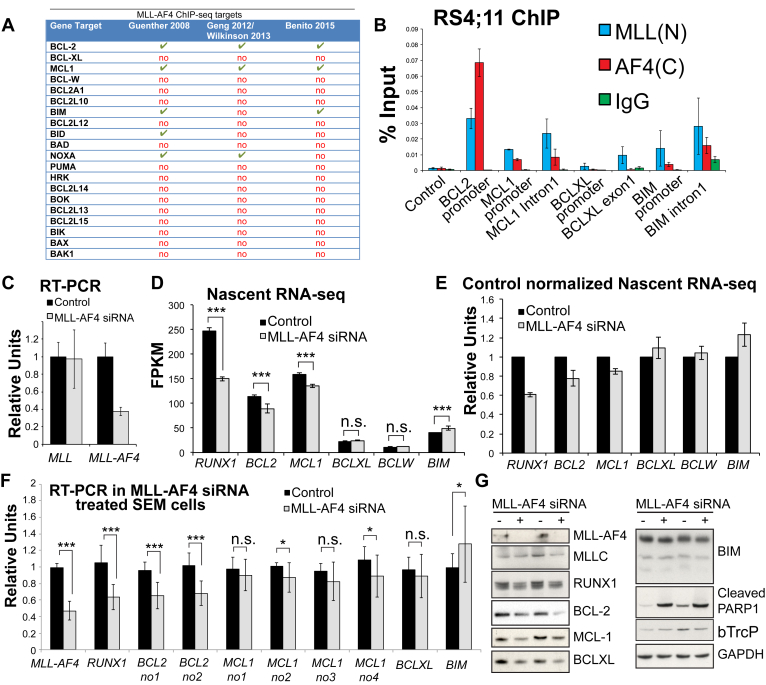
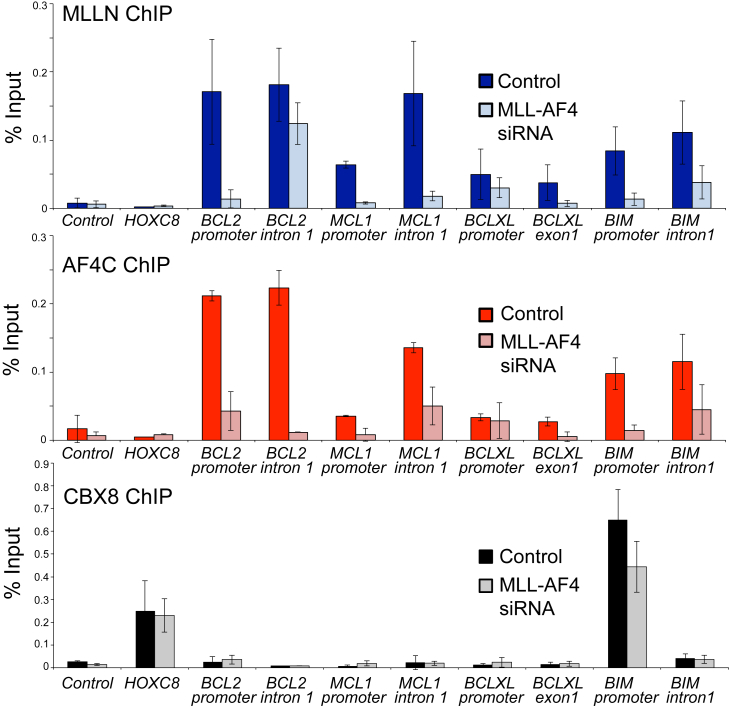
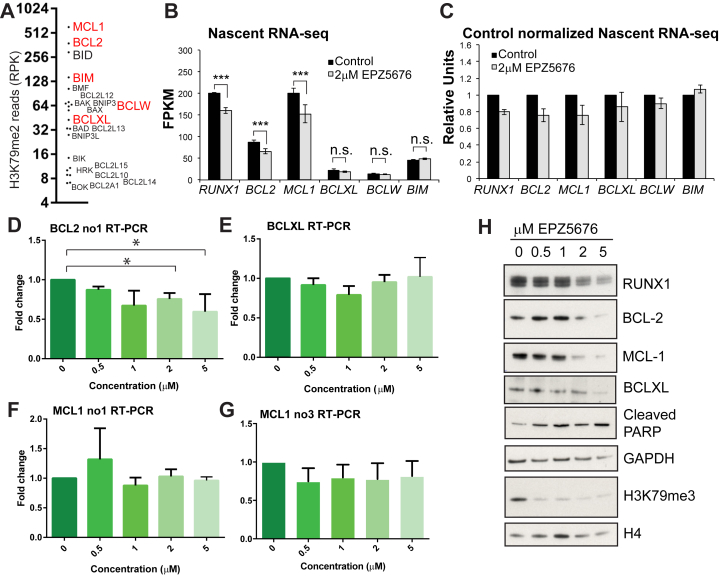
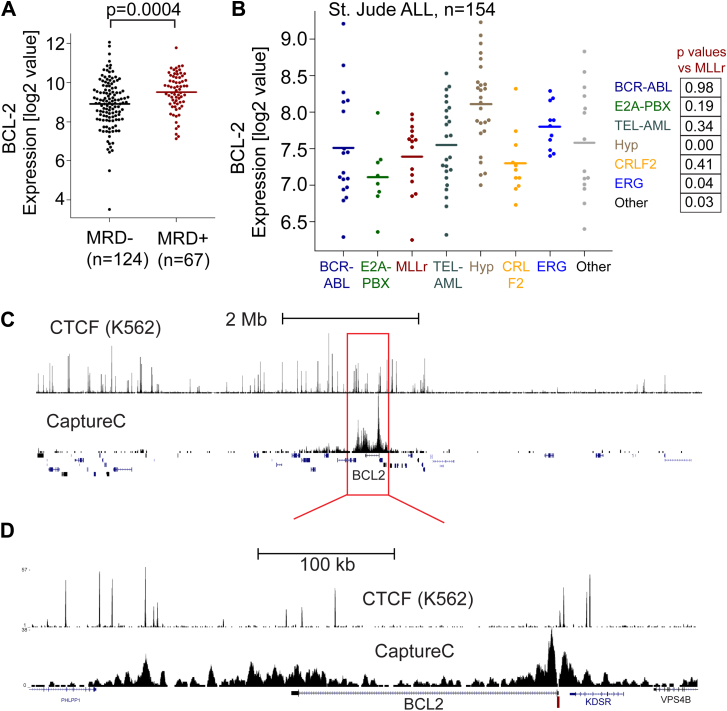
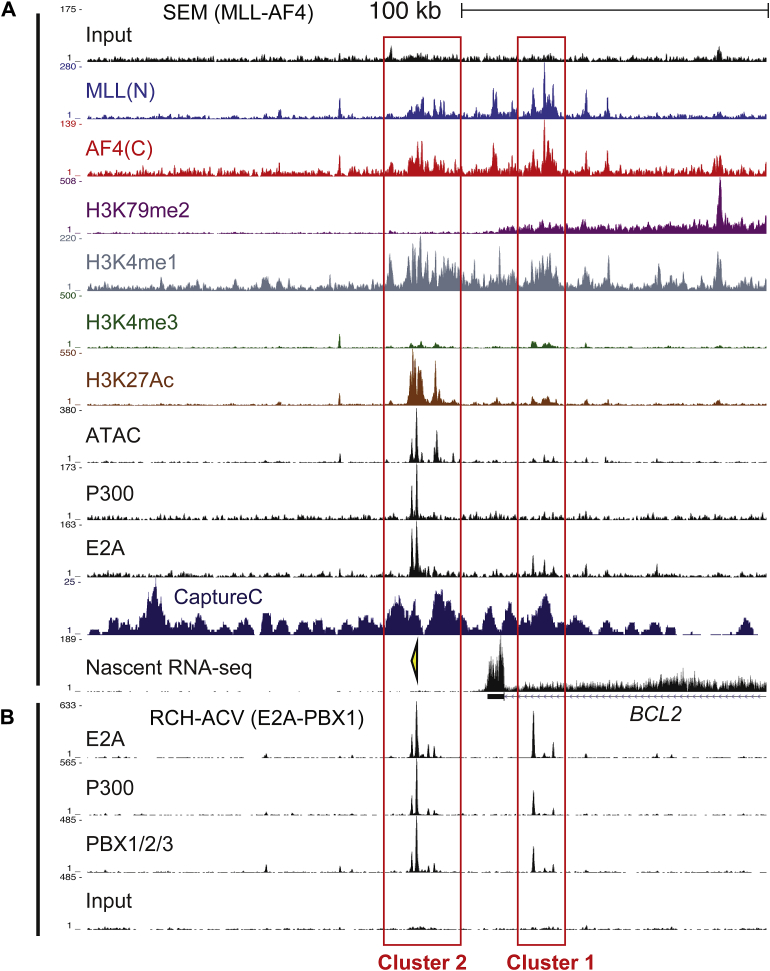
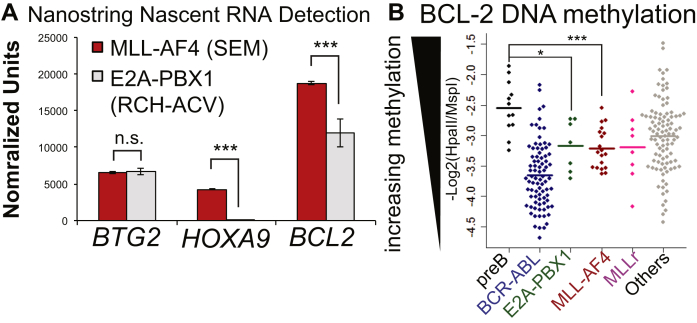
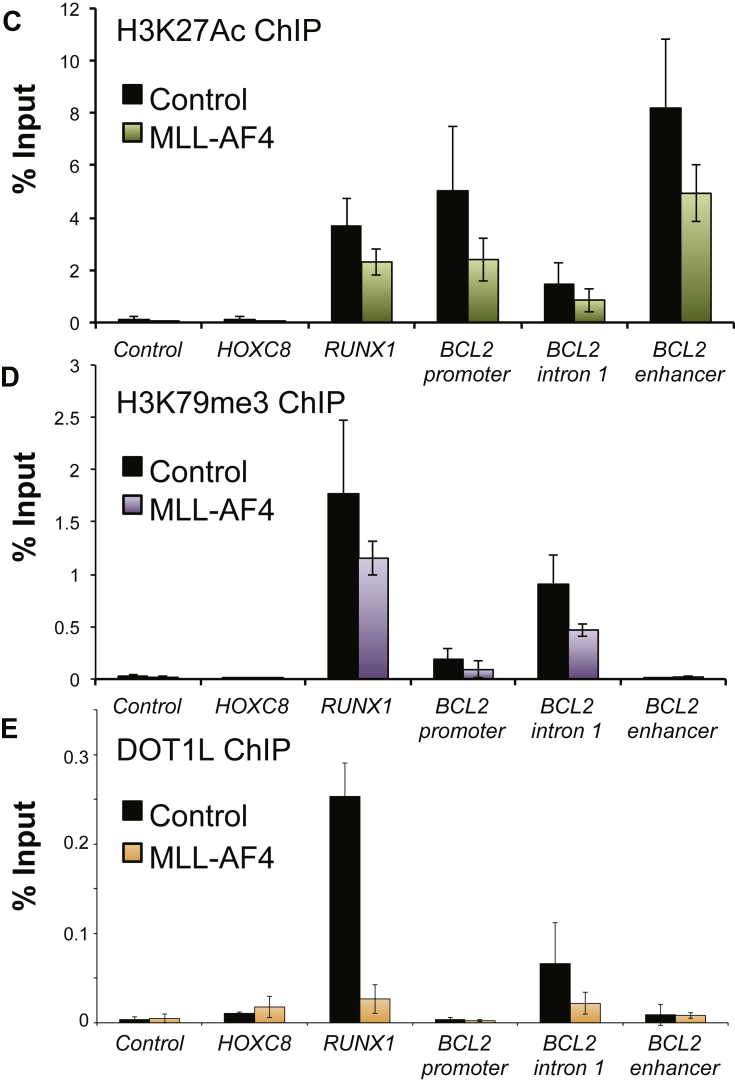
Survival rates for children and adults carrying mutations in the Mixed Lineage Leukemia (MLL) gene continue to have a very poor prognosis. The most common MLL mutation in acute lymphoblastic leukemia is the t(4;11)(q21;q23) chromosome translocation that fuses MLL in-frame with the AF4 gene producing MLL-AF4 and AF4-MLL fusion proteins. Previously, we found that MLL-AF4 binds to the BCL-2 gene and directly activates it through DOT1L recruitment and increased H3K79me2/3 levels. In the study described here, we performed a detailed analysis of MLL-AF4 regulation of the entire BCL-2 family. By measuring nascent RNA production in MLL-AF4 knockdowns, we found that of all the BCL-2 family genes, MLL-AF4 directly controls the active transcription of both BCL-2 and MCL-1 and also represses BIM via binding of the polycomb group repressor 1 (PRC1) complex component CBX8. We further analyzed MLL-AF4 activation of the BCL-2 gene using Capture-C and identified a BCL-2-specific enhancer, consisting of two clusters of H3K27Ac at the 3' end of the gene. Loss of MLL-AF4 activity results in a reduction of H3K79me3 levels in the gene body and H3K27Ac levels at the 3' BCL-2 enhancer, revealing a novel regulatory link between these two histone marks and MLL-AF4-mediated activation of BCL-2.
Copyright © 2016 ISEH - International Society for Experimental Hematology. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
