

Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism
- PMID: 27858759
- PMCID: PMC5240594
- DOI: 10.3233/JND-150088
Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism
Abstract
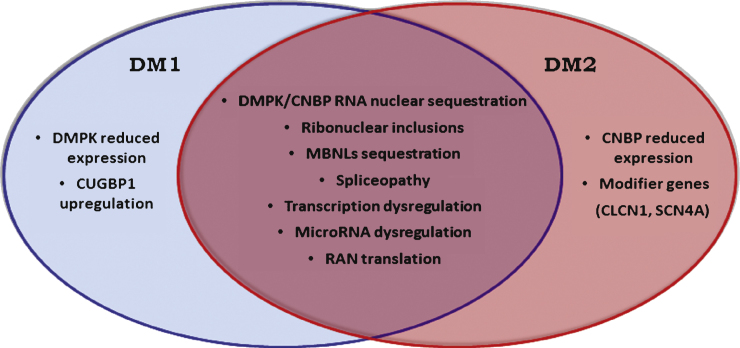
Myotonic dystrophy (DM) is the most common adult muscular dystrophy, characterized by autosomal dominant progressive myopathy, myotonia and multiorgan involvement. To date two distinct forms caused by similar mutations have been identified. Myotonic dystrophy type 1 (DM1, Steinert's disease) is caused by a (CTG)n expansion in DMPK, while myotonic dystrophy type 2 (DM2) is caused by a (CCTG)n expansion in CNBP. Despite clinical and genetic similarities, DM1 and DM2 are distinct disorders. The pathogenesis of DM is explained by a common RNA gain-of-function mechanism in which the CUG and CCUG repeats alter cellular function, including alternative splicing of various genes. However additional pathogenic mechanism like changes in gene expression, modifier genes, protein translation and micro-RNA metabolism may also contribute to disease pathology and to clarify the phenotypic differences between these two types of myotonic dystrophies.This review is an update on the latest findings specific to DM2, including explanations for the differences in clinical manifestations and pathophysiology between the two forms of myotonic dystrophies.
Keywords: Myotonic dystrophy type 1; clinical findings; molecular mechanism; muscle biopsy; myotonic dystrophy type 2; pathology.
Figures

References
-
- Harper PS. 3rd edition. Saunders; London: 2001. Myotonic Dystrophy.
-
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hutnter K, Stanton VP, Thirion JP, Hudson T. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. - PubMed
-
- Fu YH, Pizzuti A, Fenwick RG, Jr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. - PubMed
-
- Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O’Hoy K. Myotonic dystrophy mutation: An unstable CTG repeat in the 3’ untranslated region of the gene. Science. 1992;255:1253–1255. - PubMed
-
- Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, Moxley RT., 3rd Proximal myotonic myopathy: A new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44:1448–1452. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
