

Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies
- PMID: 27860548
- PMCID: PMC11060135
- DOI: 10.1146/annurev-pharmtox-010715-103335
Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies
Abstract
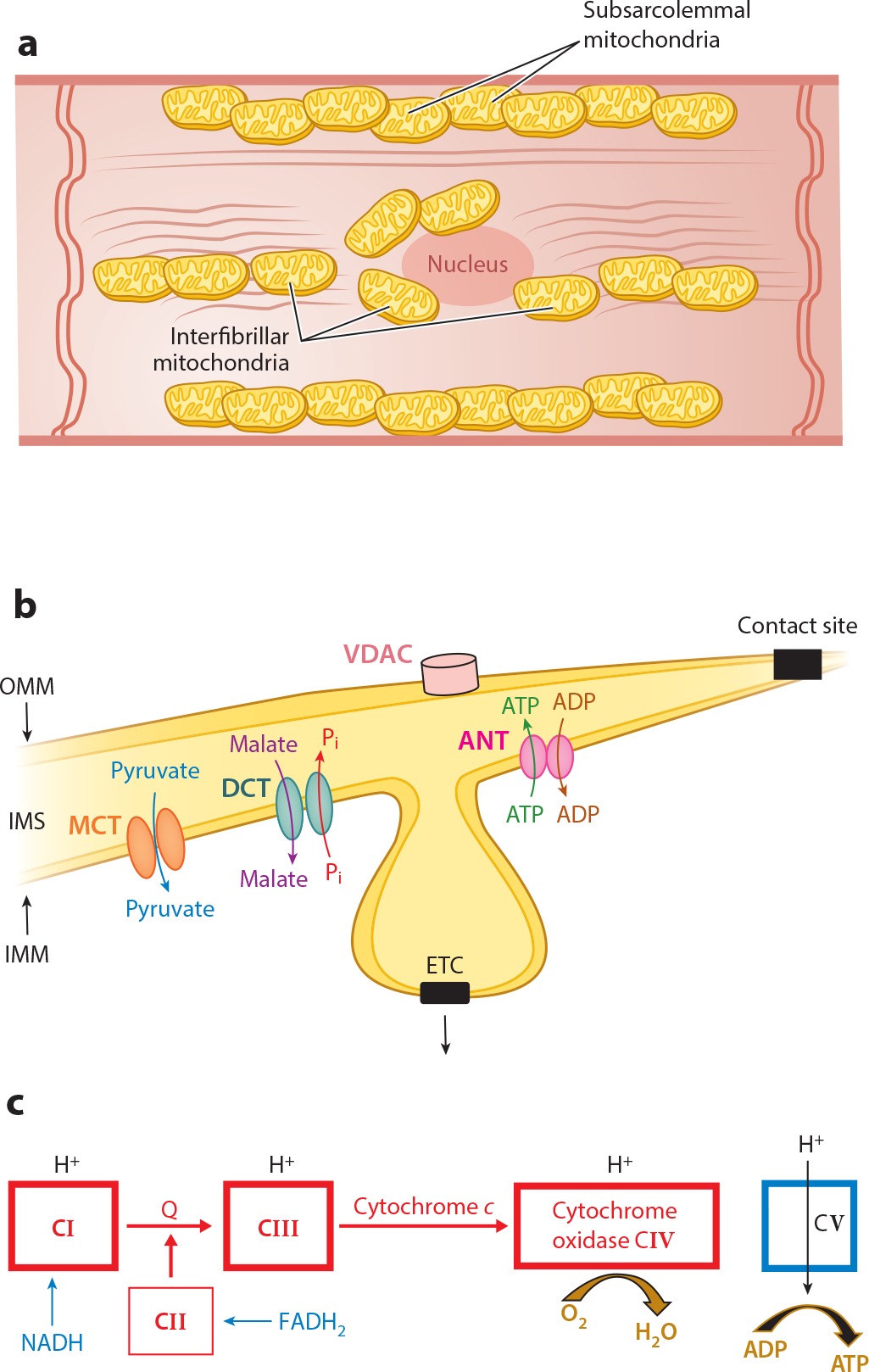
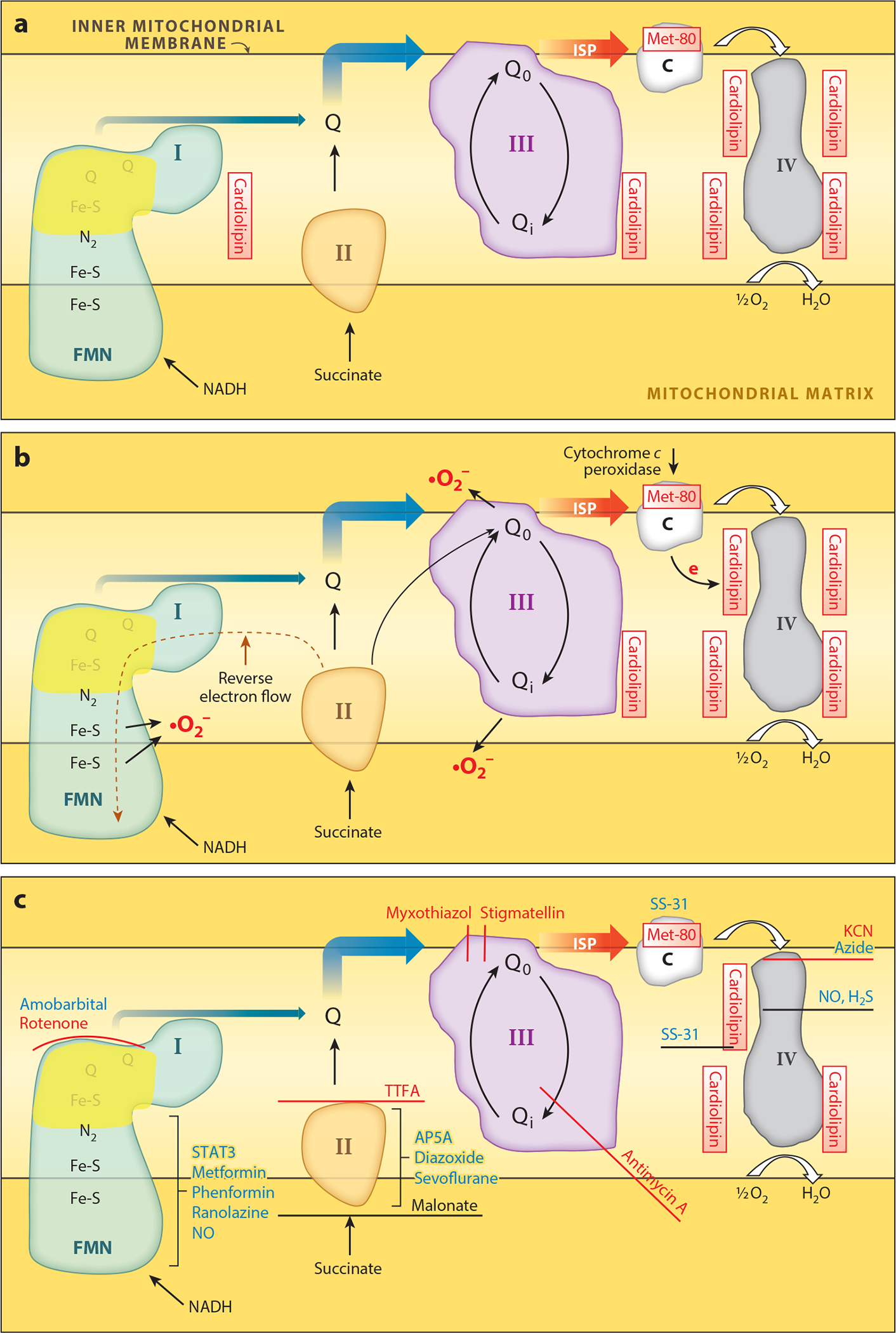
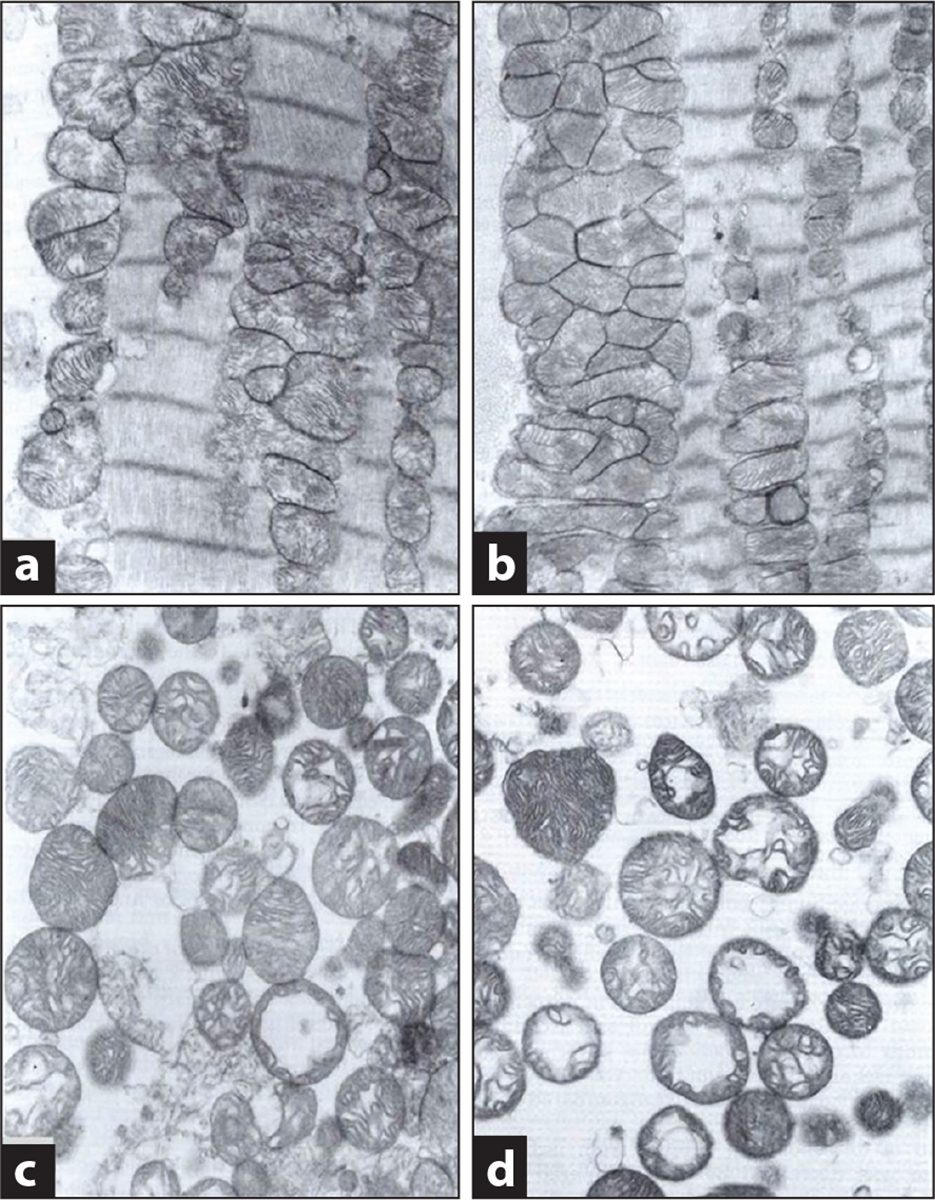
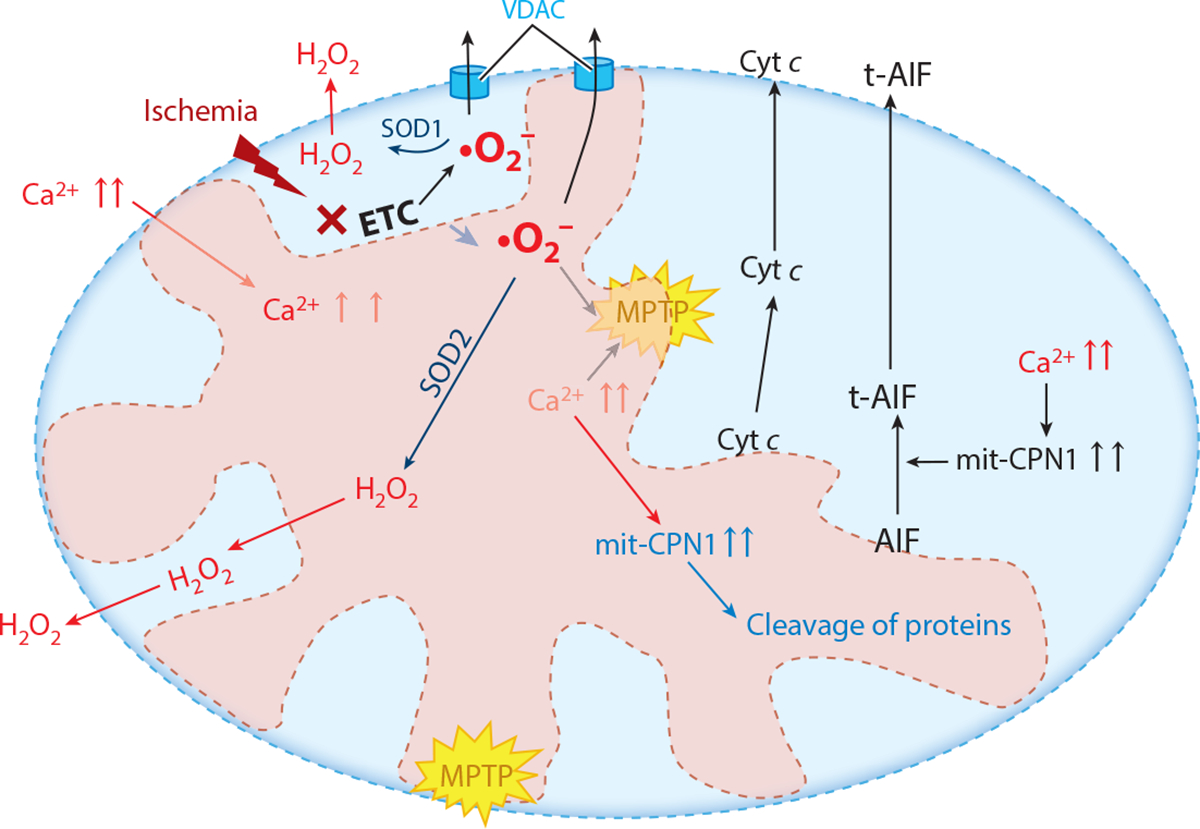
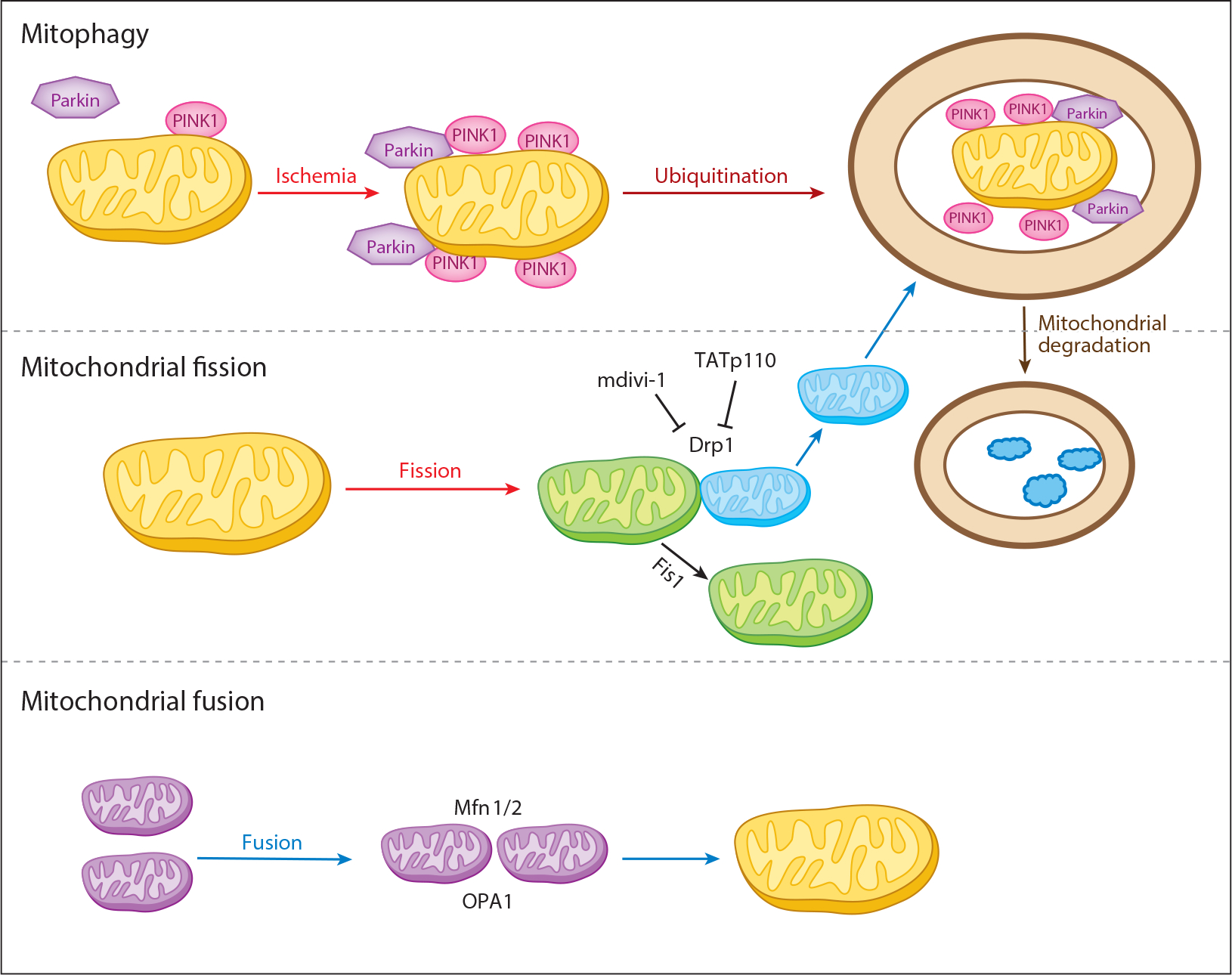
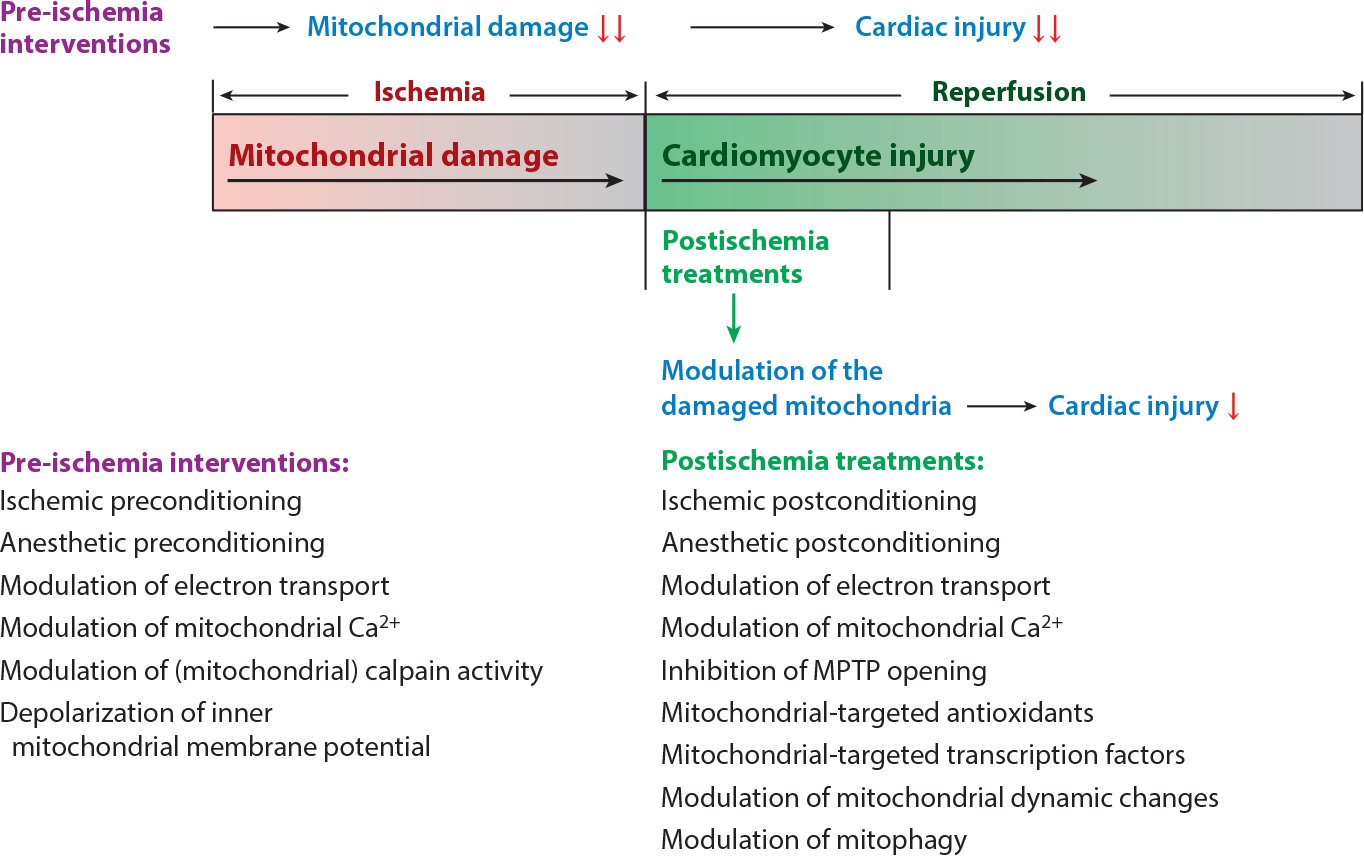
Mitochondria have emerged as key participants in and regulators of myocardial injury during ischemia and reperfusion. This review examines the sites of damage to cardiac mitochondria during ischemia and focuses on the impact of these defects. The concept that mitochondrial damage during ischemia leads to cardiac injury during reperfusion is addressed. The mechanisms that translate ischemic mitochondrial injury into cellular damage, during both ischemia and early reperfusion, are examined. Next, we discuss strategies that modulate and counteract these mechanisms of mitochondrial-driven injury. The new concept that mitochondria are not merely stochastic sites of oxidative and calcium-mediated injury but that they activate cellular responses of mitochondrial remodeling and cellular reactions that modulate the balance between cell death and recovery is reviewed, and the therapeutic implications of this concept are discussed.
Keywords: cardiolipin; electron transport chain; fatty acid oxidation; oxidative phosphorylation; reactive oxygen species; ubiquinol:cytochrome c oxidoreductase.
Figures
References
-
- Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. 2007. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Cell Physiol. 292:C137–47 - PubMed
-
- Yellon DM, Hausenloy DJ. 2007. Myocardial reperfusion injury. N. Engl. J. Med. 357:1121–35 - PubMed
-
- Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. 1977. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size versus duration of coronary occlusion in dogs. Circulation 56:786–94 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
