

Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways
- PMID: 27861609
- PMCID: PMC5115767
- DOI: 10.1371/journal.pone.0166583
Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways
Abstract
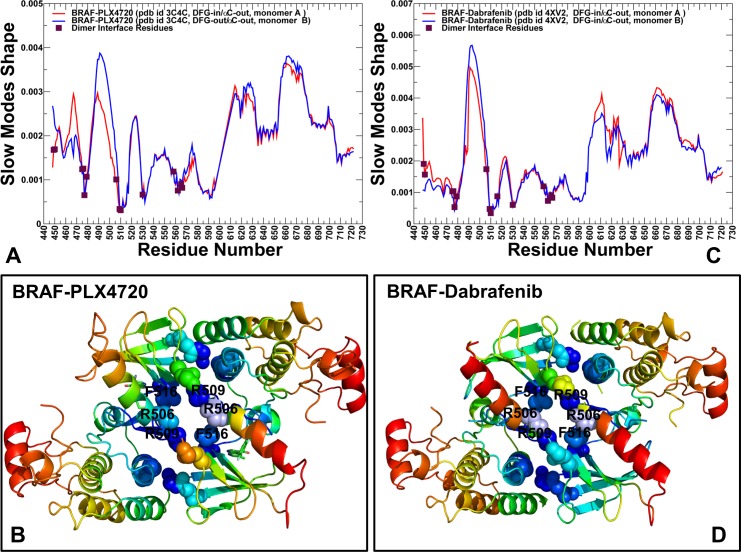
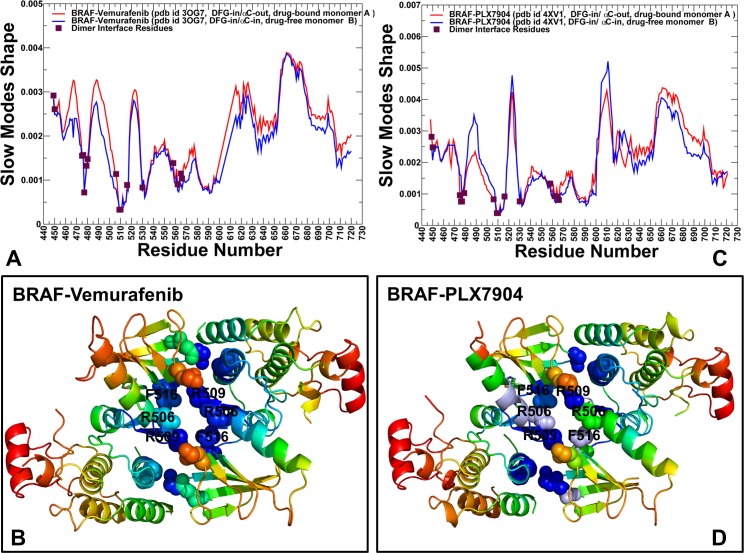
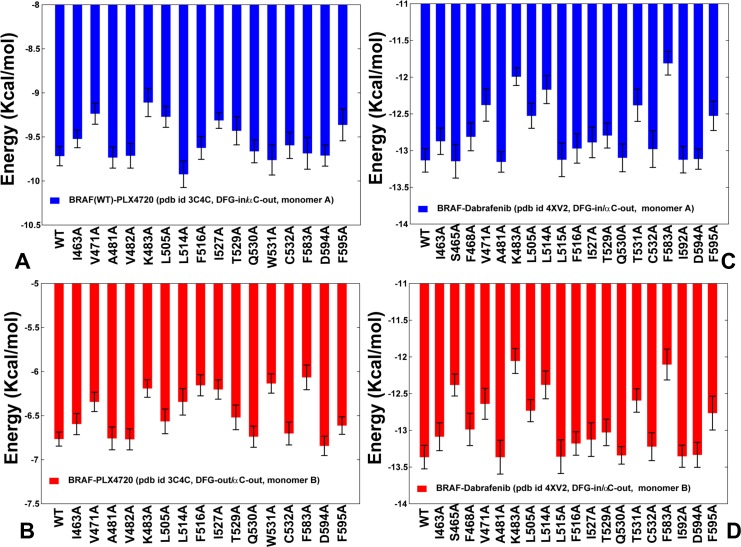
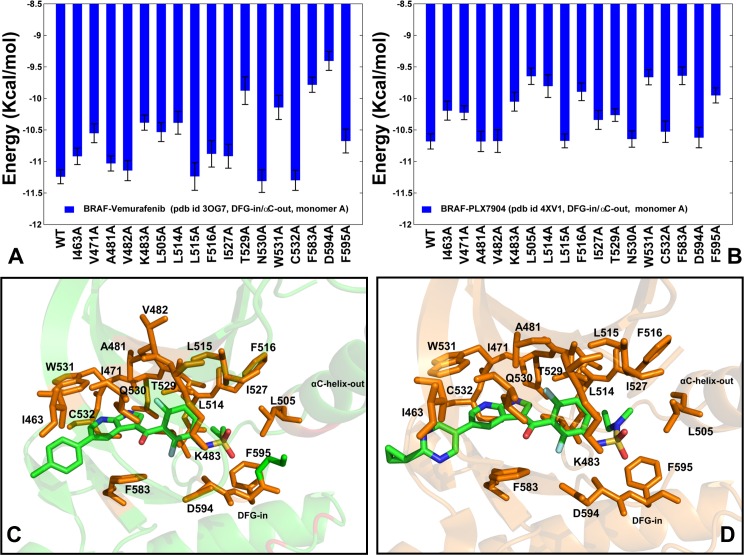
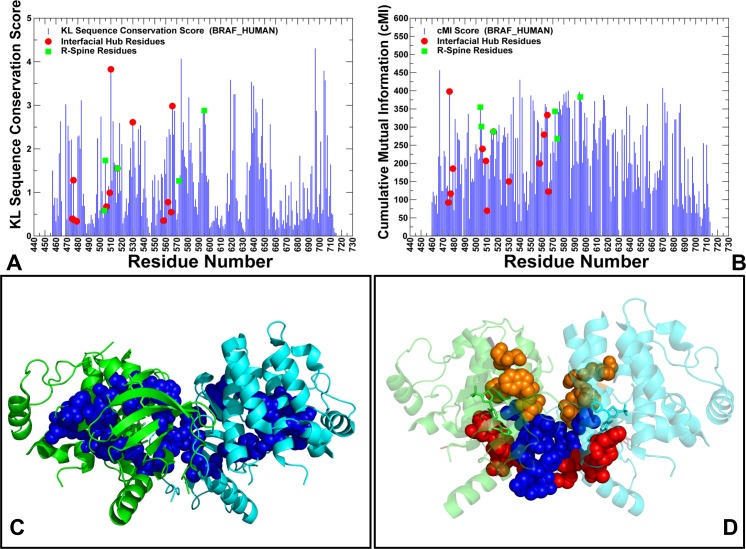
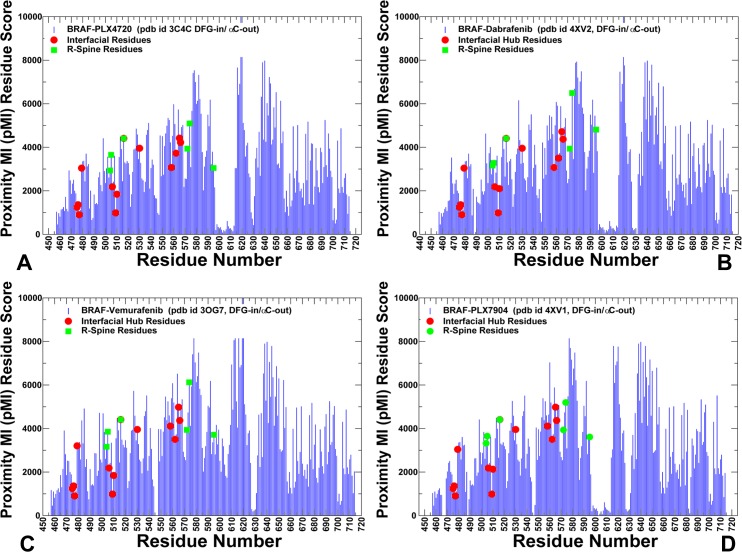
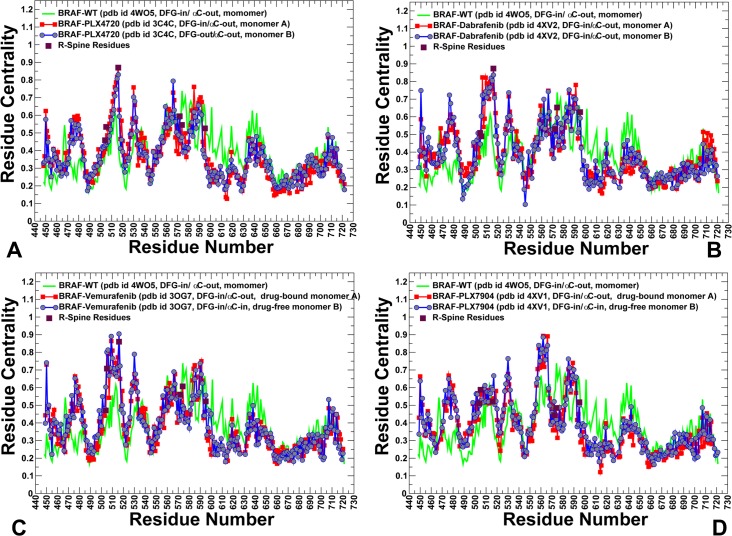
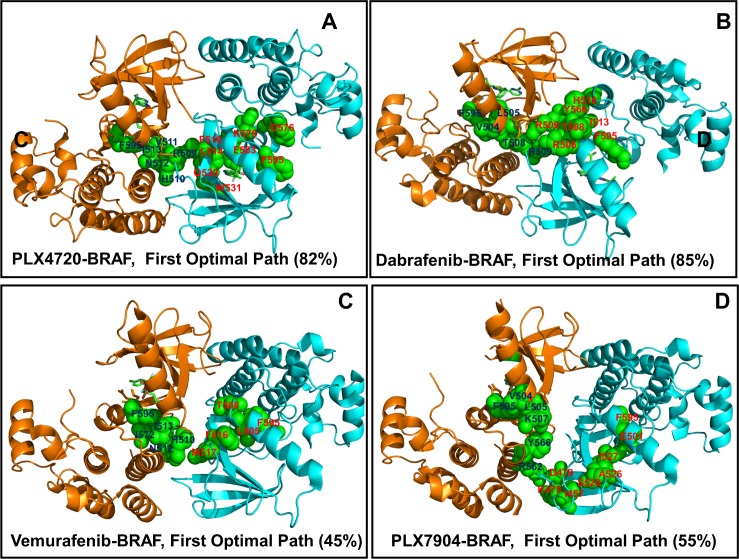
The recent studies have revealed that most BRAF inhibitors can paradoxically induce kinase activation by promoting dimerization and enzyme transactivation. Despite rapidly growing number of structural and functional studies about the BRAF dimer complexes, the molecular basis of paradoxical activation phenomenon is poorly understood and remains largely hypothetical. In this work, we have explored the relationships between inhibitor binding, protein dynamics and allosteric signaling in the BRAF dimers using a network-centric approach. Using this theoretical framework, we have combined molecular dynamics simulations with coevolutionary analysis and modeling of the residue interaction networks to determine molecular determinants of paradoxical activation. We have investigated functional effects produced by paradox inducer inhibitors PLX4720, Dabrafenib, Vemurafenib and a paradox breaker inhibitor PLX7904. Functional dynamics and binding free energy analyses of the BRAF dimer complexes have suggested that negative cooperativity effect and dimer-promoting potential of the inhibitors could be important drivers of paradoxical activation. We have introduced a protein structure network model in which coevolutionary residue dependencies and dynamic maps of residue correlations are integrated in the construction and analysis of the residue interaction networks. The results have shown that coevolutionary residues in the BRAF structures could assemble into independent structural modules and form a global interaction network that may promote dimerization. We have also found that BRAF inhibitors could modulate centrality and communication propensities of global mediating centers in the residue interaction networks. By simulating allosteric communication pathways in the BRAF structures, we have determined that paradox inducer and breaker inhibitors may activate specific signaling routes that correlate with the extent of paradoxical activation. While paradox inducer inhibitors may facilitate a rapid and efficient communication via an optimal single pathway, the paradox breaker may induce a broader ensemble of suboptimal and less efficient communication routes. The central finding of our study is that paradox breaker PLX7904 could mimic structural, dynamic and network features of the inactive BRAF-WT monomer that may be required for evading paradoxical activation. The results of this study rationalize the existing structure-functional experiments by offering a network-centric rationale of the paradoxical activation phenomenon. We argue that BRAF inhibitors that amplify dynamic features of the inactive BRAF-WT monomer and intervene with the allosteric interaction networks may serve as effective paradox breakers in cellular environment.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




Similar articles
-
Molecular dynamics simulations and modelling of the residue interaction networks in the BRAF kinase complexes with small molecule inhibitors: probing the allosteric effects of ligand-induced kinase dimerization and paradoxical activation.Mol Biosyst. 2016 Oct 20;12(10):3146-65. doi: 10.1039/c6mb00298f. Epub 2016 Aug 2. Mol Biosyst. 2016. PMID: 27481329
-
Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication.PLoS Comput Biol. 2017 Jan 17;13(1):e1005299. doi: 10.1371/journal.pcbi.1005299. eCollection 2017 Jan. PLoS Comput Biol. 2017. PMID: 28095400 Free PMC article.
-
Allosteric coupling asymmetry mediates paradoxical activation of BRAF by type II inhibitors.Elife. 2024 May 14;13:RP95481. doi: 10.7554/eLife.95481. Elife. 2024. PMID: 38742856 Free PMC article.
-
Leveraging Structural Diversity and Allosteric Regulatory Mechanisms of Protein Kinases in the Discovery of Small Molecule Inhibitors.Curr Med Chem. 2017;24(42):4838-4872. doi: 10.2174/0929867323666161006113418. Curr Med Chem. 2017. PMID: 27719654 Review.
-
Inhibition of RAF dimers: it takes two to tango.Biochem Soc Trans. 2021 Feb 26;49(1):237-251. doi: 10.1042/BST20200485. Biochem Soc Trans. 2021. PMID: 33367512 Free PMC article. Review.
Cited by
-
The structural basis for Ras activation of PI3Kα lipid kinase.Phys Chem Chem Phys. 2019 Jun 5;21(22):12021-12028. doi: 10.1039/c9cp00101h. Phys Chem Chem Phys. 2019. PMID: 31135801 Free PMC article.
-
ProSNEx: a web-based application for exploration and analysis of protein structures using network formalism.Nucleic Acids Res. 2019 Jul 2;47(W1):W471-W476. doi: 10.1093/nar/gkz390. Nucleic Acids Res. 2019. PMID: 31114881 Free PMC article.
-
A Secondary Mutation in BRAF Confers Resistance to RAF Inhibition in a BRAFV600E-Mutant Brain Tumor.Cancer Discov. 2018 Sep;8(9):1130-1141. doi: 10.1158/2159-8290.CD-17-1263. Epub 2018 Jun 7. Cancer Discov. 2018. PMID: 29880583 Free PMC article.
-
Mechanism of dimer selectivity and binding cooperativity of BRAF inhibitors.Elife. 2025 Feb 13;13:RP95334. doi: 10.7554/eLife.95334. Elife. 2025. PMID: 39945510 Free PMC article.
-
Targeted mass spectrometry-based assays enable multiplex quantification of receptor tyrosine kinase, MAP Kinase, and AKT signaling.Cell Rep Methods. 2021 Jul 26;1(3):100015. doi: 10.1016/j.crmeth.2021.100015. Epub 2021 Jun 14. Cell Rep Methods. 2021. PMID: 34671754 Free PMC article.
References
-
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109: 275–282. - PubMed
-
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;;15: 661–675. - PubMed
-
- Endicott JA, Noble ME, Johnson LN. The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem. 2012;;81: 587–613. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
